Kasuistik
En sjælden form for svær overvægt hos børn og unge
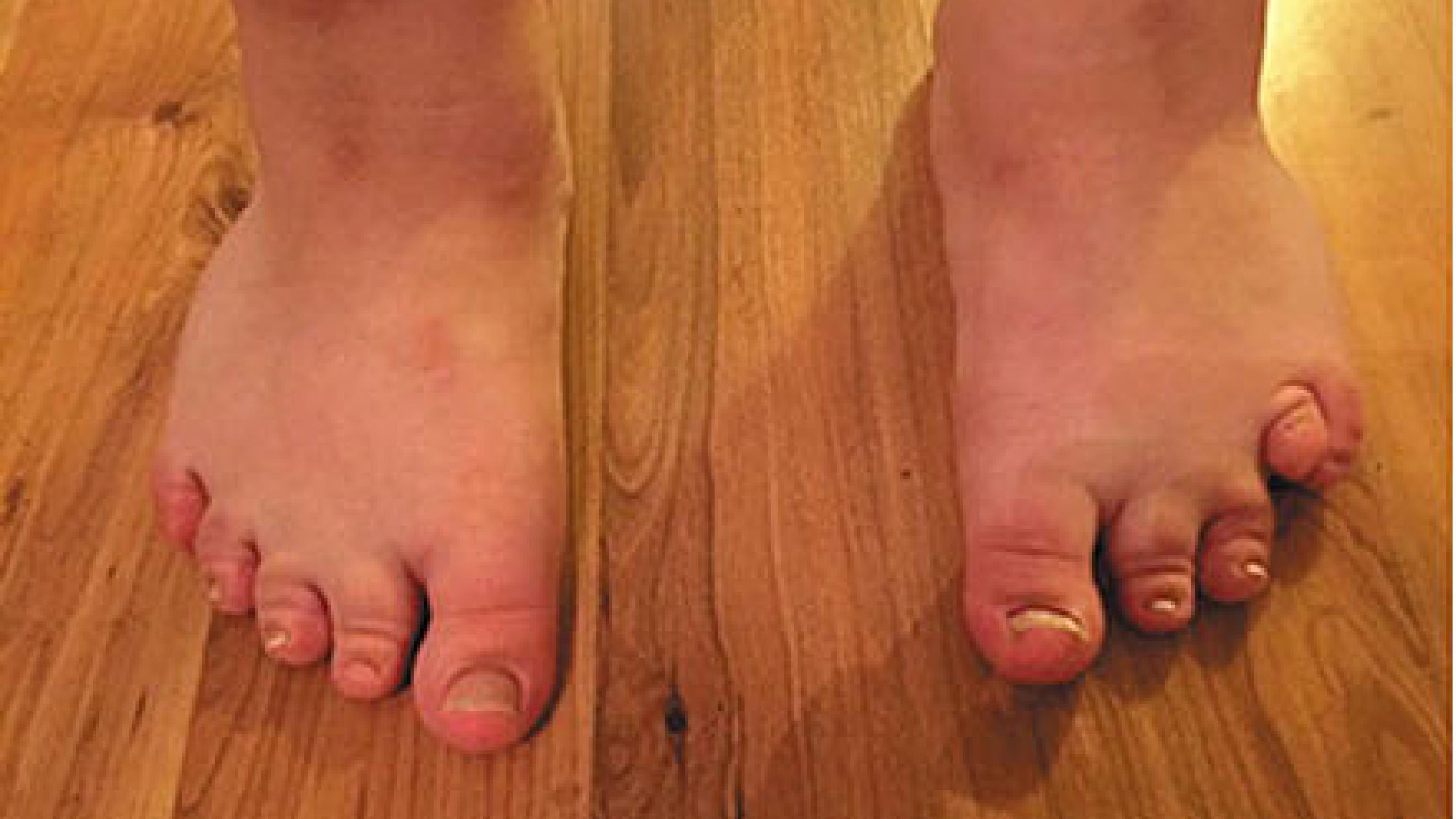
På billedet ses en karakteristisk kort fjerde tå (brakydaktyli) på venstre fod hos sygehistoriens patient med Albrights hereditære osteodystrofi.
20. feb. 2017
4 min.
Mindre end 1% af alle børn med svær overvægt har endokrine lidelser [1]. Årsagerne til lille højde kan være flere [2]. Ved Albrights hereditære osteodystrofi (AHO) er begge symptomer til stede [3]. Sygehistorien handler om en fireårig pige, der blev henvist pga. lille højde, overvægt og hypotyreose. En karakteristisk paraklinik med bl.a. forhøjet niveau af P- parathyroideahormon (PTH), hypotyreose, væksthormonmangel samt påfaldende fænotype gav mistanke om AOH, og diagnosen blev verificeret ved en molekylærgenetisk test.
En tidligere rask fire år gammel pige uden endokrin disposition blev henvist pga. lille højde, vægtøgning
og forhøjet niveau af thyroideastimulerende hormon (TSH). Udviklingsmæssigt var hun lidt forsinket.
Niveauet af de perifere thyroideahormoner var normalt, men pga. vedvarende forhøjet TSH-niveau på 9,3 mE/l (normalværdi: 0,3-4 mE/l), lav væksthastighed, overvægt og knoglealder, der var 1,3 år forsinket, blev pigen sat i behandling med levothyroxin og henvist til en diætist.
Som seksårig var pigen eutyroid, og vægten var stabiliseret, men væksthastigheden var fortsat lav. Knoglealderen var dissocieret mellem 3,5 og 6 år. To væksthormonstimulationstest viste partiel væksthormonmangel med peak-værdierne 2,4 og 5,8 mE/l (normalværdi > 15 mE/l), og hun blev sat i væksthormonbehandling. Resultaterne af MR-skanning af cerebrum, karyotypebestemmelse og array-komparativ genomisk hybridisering var normale.
Over de næste år fik hun en massiv vægtøgning (Figur 1), begyndte i specialklasse og blev flere gange indlagt med kramper i benene samt fokale og generaliserede kramper. Der begyndte at vise sig et billede med forsinket psykomotorisk udvikling og fænotypisk syndromsuspekt udseende med bl.a. et stort underbid. Røntgenbilleder viste korte fingre og tæer, især fjerde og femte stråle. I forbindelse med febrile infektioner blev pigen flere gange indlagt med muskelkramper, hvor P-PTH-niveauet var forhøjet til 25,3 og 21,9 pmol/l (normalværdi: 1,6-6,9 pmol/l). Kun en enkelt gang var niveauet af ioniseret calcium marginalt for lavt på 1,10 mmol/l (normalværdi: 1,18-1,32 mmol/l), ellers var det normalt. P-fosfatniveauet lå højt i normalområdet 1,81 mmol/l (normalværdi: 1,16-1,81 mmol/l) og P-25-OH-D-vitaminniveau var lavt på
51, 48 nmol/l (normalværdi > 50 nmol/l).
På mistanke om AOH blev patienten henvist til Center for Sjældne Sygdomme på Rigshospitalet. Mutationsundersøgelsen i GNAS bekræftede diagnosen og viste to variationer i heterozygot form, c.1154G>A, p.Arg385His. Endvidere fandt man variationen c.1036C>G p.Leu346Val, der ikke tidligere er beskrevet, og betydningen er ikke kendt. I en alder af 16 år var pigen massivt overvægtig (BMI 36,8 kg/m2) og var 159,6 cm høj. Hun blev behandlet med alfacalcidol, væksthormon, levothyroxin og levetiracetam og fulgt i børneendokrinologisk ambulatorium, overvægtsambulatorium, børne-ungdomspsykiatrisk ambulatorium, Odontologisk Videnscenter og Center for Sjældne Sygdomme. Hun gik i specialskole og havde en støttekontaktperson samt en aflastningsfamilie.
Lille højde og overvægt er hyppige pædiatriske henvisningsårsager [1, 2], men AHO er en sjælden årsag. AHO er en sjælden arvelig sygdom med en prævalens på 1,1 pr. 100.000 personer i Danmark [4].
Mutationen sidder i GNAS på kromosom 20 eller er spontant opstået. Sygdomsgenets cytogenetiske lokalisation er 20q13.32. Arvegangen er dominant arvelig [5], men fænotypen afhænger af, hvilken af forældrene der giver sygdomsmutationen videre. Når mutationen er på den allel, der er nedarvet maternelt, vil barnet udvikle en type af AHO med hormonel resistens kaldet pseudohypoparatyroidisme type 1A (PHPIa). Det er den undertype patienten i sygehistorien havde.
Hvis mutationen derimod er lokaliseret på den paternelt nedarvede allel, vil barnet få AHO uden endokrinologiske problemer (pseudopseudohypoparatyrodisme). Denne forskel opstår pga. imprintning, dvs. inaktivering af en allel, hvilket medfører forskellig genekspression, afhængig af hvilken af forældrene der bærer mutationen. PHPIa skyldes således, at der kun dannes alfaprotein fra den paternelle allel, hvilket medfører nedsat aktivering af adenylat-cyklase-cyklisk adenosinmonofosfat-systemet og dermed lavt niveau af cyklisk adenosinmonofosfat.
Det leder til, at PTH ikke kan udøve sin virkning på målorganerne pga. ændret PTH-post-receptor-signalvej [5]. Det påvirker renale tubuli og leder til hypokalcæmi, hyperfosfatæmi samt sekundær hyperparatyroidisme. GNAS findes også i bl.a. glandula thyroidea, og patienter med undertype 1 kan derfor have resistens over for andre g-koblede proteiner herunder TSH-receptoren [5].
Selv om patienten i sygehistorien ikke havde den karakteristiske hypokalcæmi, der ofte er til stede ved AHO, var de øvrige parakliniske og kliniske fund så karakteristiske, at der blev undersøgt for AHO. Ved AHO kan brakydaktyli, lav højde, overvægt, et rundt ansigt, små forkalkninger i underhuden, tandproblemer og mental retardering forekomme.
Korrespondance: Sara Østergaard Christensen.
E-mail: saraoestergaard@hotmail.com
Antaget: 13. december 2016
Publiceret på Ugeskriftet.dk: 20. februar 2017
Interessekonflikter: ingen.
A four-year-old girl was referred to a paediatric department with low height, obesity and hypothyroidism. Her paraclinical tests were characteristic with elevated P-parathyroid hor­mone concentration, hypothyroidism, growth hormone deficiency, abnormal phenotype with brachydactyly, tooth problems and mental retardation, which led to a suspicion of Albright’s hereditary osteodystrophy (AHO). The diagnosis was verified by molecular genetic testing. Less than 1% of children with obesity have an endocrine disorder, and AHO is one of them.
Gurnani M, Birken C, Hamilton J. Childhood obesity: causes, consequences, and management? Pediatr Clin North Am 2015;62:821-40.
Rogol AD, Hayden GF. Etiologies and early diagnosis of short stature and growth failure in children and adolescents. J Pediatr 2014;164
(suppl 5):1-14.e6.
Wilson L, Trembath R. Albright‘s hereditary osteodystrophy. J Med Genet 1994;31:779-84.
Underbjerg L, Sikjaer T, Mosekilde L et al. Pseudohypoparathyroidism – epidemiology, mortality and risk of complications. Clin Endocrinol 2015;
84:904-11.
Mantovani G. Clinical review: pseudohypoparathyroidism: diagnosis and treatment. J Clin Endocrinol Metab 2011;96:3020-30.