Invited state-of-the-art review
Genetics of metabolic traits in Greenlanders
16. nov. 2023
17 min.
Abstract
The history of the Greenlandic population has contributed to a unique genetic composition in which high-impact and often novel genetic variants segregate at a high frequency. As a result, research in smaller populations like the Greenlandic has the potential to reveal genetic variation and associations that are not present or discoverable in other populations. In this review, we provide a summary of our research in Greenlanders and our findings of genetic variants with high impact on metabolic health, and outline the implications that this research has for the equitable distribution of genomic precision medicine.
Key Points
The history of the Greenlandic population has contributed to establishing a unique genetic architecture.
Several novel variants associated with metabolic traits have been identified in Greenlanders. These include high-impact variants, which may potentially be of clinical relevance or reveal novel drug targets.
Efforts are needed to ensure an equitable distribution of and access to resources that facilitate genomic precision medicine, especially in populations enriched for common high-impact variants.
Health and disease are determined by genetic and environmental factors that are shaped by our individual, familial and broader communal characteristics. Differences in genetic architecture contribute to differences in disease susceptibility between populations and are important to consider to achieve equitable access to genomic precision medicine. In this review, we summarise our findings on the genetic contribution to metabolic disease in Greenland and the perspectives that these findings hold for our understanding and treatment of metabolic disease.
The genetic architecture of the present-day Greenlandic Inuit population is shaped by the demographic history of their ancestral population. Genetic evidence indicates that the population diverged from the Han Chinese > 20,000 years ago [1] and migrated through Siberia and across the Bering Strait to the North American Arctic before reaching and populating Greenland beginning in ~ 1000 CE [2]. The ancestral Greenlandic Inuit population was characterised by having undergone a severe and prolonged population bottleneck, lasting for at least 15,000 years, and by having a small effective population size with limited gene flow from other populationst are variable (i.e. depletion of rare alleles) [3, 5]. Thus, whereas the collected load of deleterious variants in an additive genetic model is similar in the Greenlandic population as in a European reference population, the proportion of deleterious variants that are common is higher in Greenland [3, 5].
These fluctuations occur in all populations; however, small populations like the Greenlandic are more prone to the effects of genetic drift as each individual represents a larger proportion of the total population. Founder effects occur when a subset of a population founds a new population, producing a genetic makeup characterised by the subset of genetic diversity in the founders, which may be different from that of the original population. In addition, there is evidence of selective adaptations to the extreme Arctic environment and a diet rich in polyunsaturated fatty acids [1]. While the majority of the descendent Greenlandic population is admixed and have both Inuit and European ancestry, admixture has occurred recently, i.e. within the past ~ 300 years [3, 4]. The small population size and historical isolation of the population has resulted in comparatively fewer variable sites but a higher allele frequency at those sites that are variable (i.e. depletion of rare alleles) [3, 5]. Thus, whereas the collected load of deleterious variants in an additive genetic model is similar in the Greenlandic population as in a European reference population, the proportion of deleterious variants that are common is higher in Greenland [3, 5].
This comparative lack of genetic diversity and consequent homogeneity in the Greenlandic population is advantageous in the context of genetic association studies because it results in less heterogeneity and improved statistical power [6, 7], and results are more likely to have broad public health implications than those of genetic discovery studies in predominantly European populations that have identified primarily low or moderate impact variants [8].
The first genome-wide association study (GWAS) of glucose-homeostasis-related traits in Greenland was proof of concept of the value of genetic association studies in small populations. Here, Moltke and colleagues analysed the genomes of 2,575 Greenlandic individuals in order to identify genetic variants that were associated with diabetes, or glucose or insulin levels at fasting or after an oral glucose load. They identified the p.R684* nonsense variant in the TBC1 domain family member 4 gene (TBC1D4), which encodes an important regulator of insulin-mediated glucose uptake in the muscle [9]. The variant was found to be strongly associated with 2-hr measures of plasma glucose levels during an oral glucose tolerance test (OGTT) and with measures of postprandial insulin resistance. On average, homozygous carriers of the p.R684* variant have almost 4 mmol/l higher blood glucose values 2 hrs after an oral glucose load than wildtype and heterozygous carriers.
Furthermore, homozygous carriers of the variant have ten-fold higher odds of developing diabetes, with more than 80% of homozygous carriers developing diabetes by 60 years of age. It was estimated that more than one in ten cases of diabetes in Greenland are attributable to this variant. While the variant is not strictly unique to the Greenlandic population, it is common only in Inuit populations, with an estimated minor allele frequency (MAF) of 23% in the Greenlandic Inuit ancestry proportion. The combination of the high effect size and high frequency means that this variant has, by far, the largest population impact on diabetes of any known diabetes variant [10] (Figure 1). Indeed, it is perhaps the only example of a common form of arguably monogenic diabetes.
The fact that the variant is associated with 2-hr but not fasting glucose values is important in the context of diagnosing diabetes. Manousaki and colleagues sequenced the variant among Canadian and Alaskan Inuit, replicating the results from the Greenlandic study [16]. They found that a diagnosis of diabetes based on fasting glucose and/or HbA1c but not based on an OGTT led to a substantial underdiagnosis of diabetes and prediabetes in the population. Fasting glucose and HbA1c only captured half of homozygous p.R684* carriers. Importantly, the authors suggested that a modified diagnostic strategy may be necessary in people of Inuit ancestry with increased use of OGTT or genetic screening for identification of diabetic individuals.
The p.R684* variant causes profound postprandial muscular insulin resistance through decreased expression and insulin-stimulated translocation of the glucose transporter GLUT4 and consequent decreased insulin-mediated glucose uptake in skeletal muscle [9]. Understanding the exact molecular basis of a disease in this manner empowers clinicians and researchers to make educated inferences about the direction for future studies and treatment opportunities. TBC1D4 plays a role in exercise-induced insulin sensitisation [17] and, therefore, the interaction between being a carrier of the TBC1D4 loss-of-function variant and exercise was of particular interest for further research. In an investigation of the interaction using data from Greenlandic population surveys, Schnurr et al. found a beneficial effect of exercise on glucose homeostasis for homozygous p.R684* variant carriers in particular [18]. They found that 2-hr blood glucose levels decreased by 0.2 mmol/l per 10 kJ/kg/day of physical activity. This means that 1 hr of high-intensity physical activity per day decreased 2-hr blood glucose by 0.7 mmol/l, a magnitude which is similar to findings from randomised controlled trials of intensive lifestyle interventions [19]. It has also been suggested that insulin-sensitising drugs of the thiazolidinedione class may be useful in treating homozygous carriers of the p.R684* variant [16]. However, whether these anti-diabetic drugs may be used to treat Greenlandic carriers of the TBC1D4 loss-of-function variant remains unknown.
While the TBC1D4 p.R684* variant was originally identified in an additive genetic model that assumes that each copy of an allele contributes proportionally to the trait, associations between genetic variants and traits may also assume a recessive genetic model where the effects of an allele are seen only when an individual carries two copies of an allele (i.e. is a homozygous carrier of a variant). In a subsequent study applying a recessive genetic model of diabetes risk, Grarup and colleagues further elucidated the genetic basis of diabetes in Greenlanders [20]. Here, they identified two additional common variants robustly associated with diabetes; one in ITGA1 (rs870992) and one near LARGE1 (rs16993330). Association analyses revealed that rs16993330 was associated with measures of abdominal fat deposition and insulin resistance, pointing towards possible mechanisms involved in the disease pathogenesis. Conversely, rs870992 was associated with HbA1c and fasting blood glucose. Neither variant was unique to the Greenlandic population; however, rs870992 was ten times and rs16993330 was more than twice as common in the Inuit ancestry component as in Europeans. In a GWAS of obesity and BMI in Greenland, Andersen et al. identified a novel variant rs4936356 near SIK3, which was associated with lower BMI, leaner body composition and improved insulin sensitivity [21]. The variant had a frequency of 28% in Inuit ancestry but only 15% in European ancestry. Thus, despite being prevalent in diverse populations, genetic discovery studies in Greenlanders have facilitated the discovery of novel genetic associations.
To detect genetic variation in a specific population, unbiased methods for discovery are occasionally required as novel and population-specific variants may exist that are not captured by, e.g., genotyping arrays. This was exemplified in the first screens of whole exome and whole genome sequence data from the Greenlandic population.
Grarup and colleagues screened whole exome data for novel loss-of-function variants in genes known to be associated with BMI or obesity, identifying the novel ADCY3 c.2433-1G>A splice variant [22] with an MAF of 3% in the Inuit ancestry component. They found that the variant had recessive effects on both body composition and diabetes risk, with homozygous carriers having 8% higher fat percentage and 2 mmol/l higher glucose values 2 hr after an oral glucose load than non-carriers. RNA sequencing in blood leukocytes showed that both heterozygous and homozygous variant carriers had decreased levels of ADCY3 mRNA. Sequencing also showed that the variant disrupts a splice-acceptor site, resulting in the processing of two alternative isoforms, with exon skipping or intron retention, respectively.
The latter was predicted to be prone to nonsense-mediated decay, further supporting ADCY3 haploinsufficiency as the causal mechanism underlying the association. Similarly, Saeed and colleagues demonstrated homozygous ADCY3 variants as a cause of monogenic obesity in the Pakistani population [23]. The mechanisms underlying the association between ADCY3 insufficiency and obesity were further elucidated by Siljee and colleagues, who showed that ADCY3 and MC4R – another gene harbouring variants associated with obesity [24] – co-localise to the primary cilia of hypothalamic neurons, and that disruption of adenylyl cyclase signalling in the primary cilia of mice increased their food intake and led to weight gain [25].
Thuesen et al. screened whole genomes from a subset of participants from Greenlandic population surveys [10] for novel genetic variation in known monogenic diabetes genes. This revealed a novel variant in HNF1A, c.1108G>T, which was associated with four-fold increased odds of diabetes, and which was functionally inferred to be splice-affecting. HNF1A is a transcription factor necessary for the transcription of proteins involved in glucose-stimulated insulin secretion [26]. In line with this, the authors observed that the insulin levels of variant carriers were more than 200 pmol/l lower 30 minutes after the ingestion of an oral-glucose load than in non-carriers, while fasting and 2-hr levels were unaffected, indicative of impaired beta-cell function. The variant was revealed to be Inuit specific, with an estimated MAF of 2% in the Inuit ancestry component.
The high impact and relative frequency of the variant means that the variant has the highest population impact on type 2 diabetes of any variant within a known monogenic diabetes gene, and is second only to the previously identified TBC1D4 loss-of-function variant (Figure 1). Collectively, almost one in five diabetes patients in Greenland are carriers of either the TBC1D4 or the HNF1A variant, demonstrating that the proportion of diabetes precipitated by high-impact variants is many-fold higher in Greenland than in larger populations. Whether carriers of this Inuit-specific HNF1A variant will benefit from treatment with sulphonylurea in the same way as people with monogenic HNF1A-diabetes [27] should be clarified in future clinical trials.
While it has traditionally been purported that Inuit have a low incidence of cardiovascular disease, more recent examinations have found that markers of cardiovascular health and the incidence of coronary heart disease are similar in Greenlanders and European populations [28-30]. Part of the explanation is rapid westernisation of diet and lifestyle [29]; however, genetic differences at the population level may also contribute to the high overall risk. Dubé and colleagues sequenced the low-density lipoprotein receptor gene (LDLR) in a sampling of Greenlandic individuals with extreme low-density lipoprotein (LDL)-cholesterol levels and identified a novel functional LDLR variant p.G137S (referred to in their manuscript using alternative nomenclature G116S). The variant was strongly associated with total and LDL-cholesterol levels. In a subsequent study, Jørsboe and colleagues characterised the variant in large Greenlandic population surveys [15]. Here, they found an MAF in the Inuit ancestry proportion of 23%, which is substantially higher than reported in previous studies. They also found a higher impact on total and LDL-cholesterol levels, with variant carriers having 0.75 mmol/l higher LDL-cholesterol levels per risk allele than non-carriers.
The combination of a high effect size and high frequency renders the Arctic LDLR variant the variant with the highest described population impact on LDL cholesterol levels (Figure 1). This was reflected in an increased risk of ischaemic heart disease, peripheral artery disease and coronary operations. The study demonstrated that the Greenlandic population may be particularly vulnerable to cardiovascular disease, particularly in light of an increasingly westernised diet and lifestyle, but that a potential exists for early prevention and treatment through identification of variant carriers.
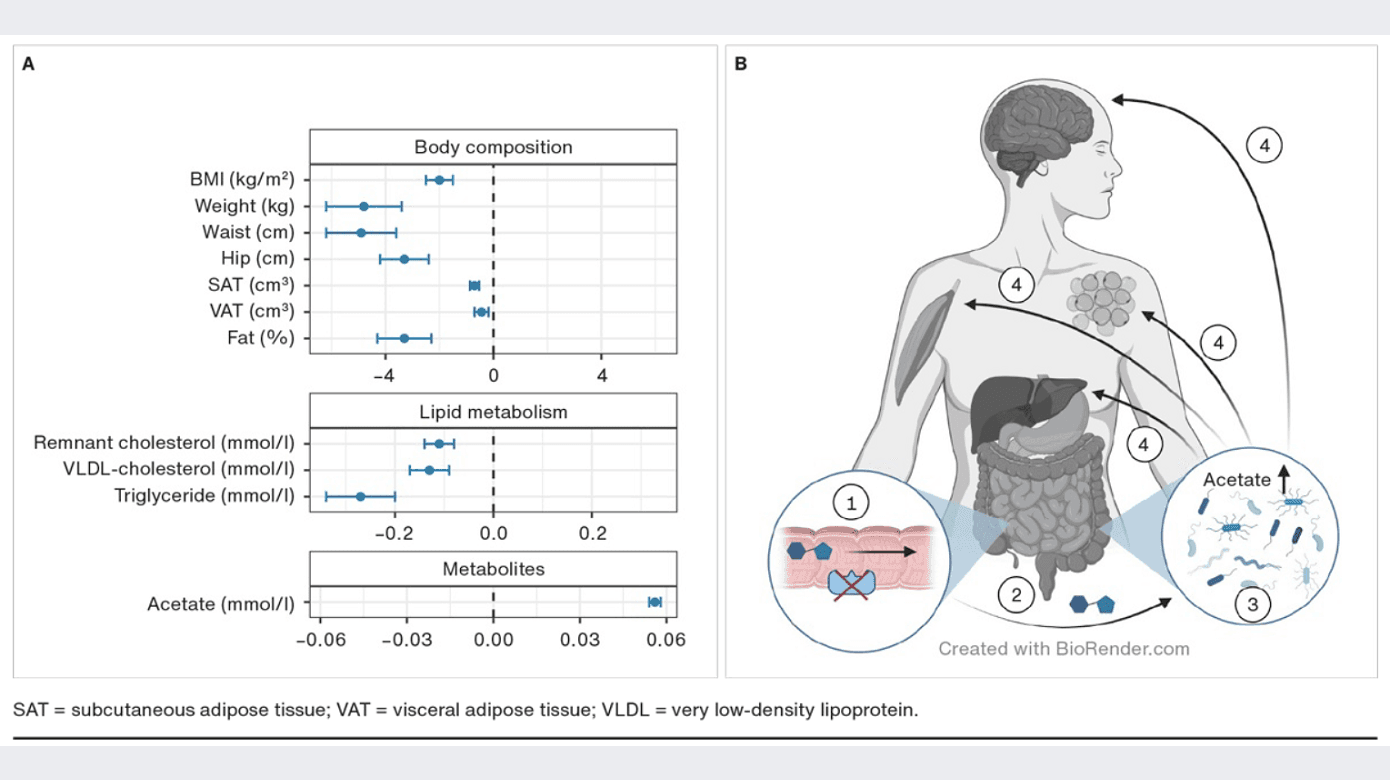
In Arctic populations, the prevalence of congenital sucrase-isomaltase deficiency is many-fold higher than in other populations, estimated at 3-4% [31]. The deficiency is characterised by gastrointestinal symptoms in childhood that usually subside by adulthood. Marcadier and colleagues were the first to identify the frameshift loss-of-function variant c.273_274delAG/p.G92Lfs in the sucrase-isomaltase gene (SI), which was found to be common in a small cohort of Canadian Inuit [32]. The SI is particularly interesting in the context of metabolic health as it cleaves carbohydrates, including sucrose to glucose and fructose, and is therefore important for sugar uptake from the small intestine. Inhibition of carbohydrate digestion is the target of antidiabetic drugs such as acarbose, which can be used to treat type 2 diabetes and prediabetes [33]. In this context, common genetic variation that completely eliminates SI function provides an opportunity for the study of the metabolic aspects of SI inhibition and elucidate SI as a potential drug target. Andersen et al. conducted a subsequent population-based study in Greenlanders, assessing the impact of the SI variant on metabolic health [34]. They found an estimated MAF of 20% in the Inuit ancestry genome, confirming that the variant is common. Homozygous carriers of the variant were markedly leaner, had a lower fat percentage and a much healthier lipid composition than heterozygous and wildtype carriers (Figure 2).
These differences were not explained by the lower intake of sugar among homozygous carriers, but rather by the higher levels of plasma acetate observed in homozygous carriers of the loss-of-function variant. The authors hypothesised that the higher levels of acetate were caused by bacterial fermentation of carbohydrates in response to loss of SI function, and that the increased acetate levels may contribute to improved metabolic health (Figure 2). The mechanisms behind the healthful effects of acetate are not fully understood. Potential pathways include increased energy expenditure, appetite regulation, increased lipid oxidation, decreased inflammation and increased thermogenesis [35, 36]. Future studies are needed to confirm these potential effects and to elucidate the potential of SI inhibition as a target for the treatment of diabetes, prediabetes and obesity.
Our work in Greenland over the past decades has revealed novel genetic variation that contributes to metabolic health in Greenlanders. Some of this variation is population-specific, occurring only in Inuit ancestry, whereas other variation is disproportionately more common in Inuit ancestry. These differences in genetic composition are precipitated by the dramatic population history of the Greenlandic Inuit. Elucidating the genetic susceptibility to disease and health in this population has contributed to revealing genetic associations, which were not discoverable in outbred populations and to identifying novel genetic variation that improves our understanding of the molecular mechanisms of disease and reveals novel targets for disease treatment and prevention. The presented studies have also provided examples of true precision medicine, where screening and treatment may be tailored by genotype (Table 1).
The disproportionate genetic contribution to metabolic health and disease in Greenland underlines the need for equitable access to genomic precision medicine. Efforts are needed to ensure that both a technical and a computational infrastructure is available in Greenland that facilitates genomic analyses. Furthermore, efforts are needed worldwide that ensure that underrepresented populations have access to genomic research as it is clear that research in populations beyond those with European ancestry are valuable both within and beyond the given population.
Correspondence Torben Hansen. E-mail: torben.hansen@sund.ku.dk
Accepted 22 September 2023
Conflicts of interest none. Disclosure forms provided by the authors are available with the article at ugeskriftet.dk/dmj
Cite this as Dan Med J 2023;70(12):A05230332
Open Access under Creative Commons License CC-BY-NC-BD 4.0
