Statusartikel
Akut eksacerbation af fibrotiske interstitielle lungesygdomme
14. aug. 2023
13 min.
Hovedbudskaber
Akut eksacerbation af idiopatisk pulmonal fibrose (AE-IPF) eller anden fibrotisk interstitiel lungesygdom er en akut og signifikant respiratorisk forværring kendetegnet ved ny og udbredt alveoleskade og høj mortalitet trods behandling.
I RCT’er af behandling af AE-IPF med hhv. cyclophosphamid eller rekombinant humant trombomodulin har man ikke kunnet påvise nogen gavnlig effekt, og behandlingen baseres derfor fortsat på glukokortikoid trods begrænset evidens.
Behandling af AE-IPF består af glukokortikoid, understøttende behandling, behandling af evt. udløsende årsag samt grundig palliativ indsats.
Idiopatisk pulmonal fibrose (IPF) er en kronisk interstitiel lungesygdom (ILS) med ukendt ætiologi, hvor den progredierende fibrosedannelse i lungerne resulterer i tiltagende respirationssvigt med restriktiv lungefunktionsnedsættelse og nedsat diffusionskapacitet [1]. Mange patienter med IPF dør af deres lungesygdom inden for få år, ofte pga. en akut eksacerbation. Også patienter med andre fibrotiske ILS end IPF (f.eks. fibrotisk allergisk alveolitis eller reumatoid artritis-associeret ILS) kan opleve akutte eksacerbationer.
En akut eksacerbation af IPF (AE-IPF) defineres som en klinisk signifikant akut respiratorisk forværring med ny og udbredt alveoleskade hos patienter med IPF [2]. Forværringen opstår typisk inden for en måned og kendetegnes ved karakteristiske nyopståede radiologiske forandringer. En AE-IPF adskiller sig væsentlig fra eksacerbationer af andre lungesygdomme som f.eks. KOL bl.a. i forhold til patofysiologi, radiologiske fund, behandling og prognose.
Incidensen af AE-IPF er usikker og afhænger af, hvilken definition der benyttes, og hvilken population der undersøges. Den estimeres at være 4-20 pr. 100 personår hos patienter med IPF [2].
Ætiologien til AE-IPF er ikke endeligt klarlagt, men AE-IPF kan være idiopatisk eller sekundær til en ekstern trigger [2]. Både virale og bakterielle infektioner kan udløse en AE-IPF, hvilket formentlig forklarer, hvorfor incidensen af eksacerbationer er højest om vinteren og foråret [2-4]. Derudover kan AE-IPF udløses af thoraxkirurgiske indgreb og procedurer (f.eks. hjertekirurgi, kirurgisk lungebiopsi eller lungecancerresektion) [5]. AE-IPF efter kirurgiske indgreb uden for thorax, efter stråleterapi eller efter invasive diagnostiske procedurer i lungerne er kasuistisk beskrevet i litteraturen [6]. En mulig forklaring på, at ekstratorakal kirurgi kan udløse en AE-IPF, er perioperativ hyperoxi eller stress af den mekanisk ventilerede lunge, evt. i kombination med et øget antal cirkulerende fibrocytter hos postoperative patienter [6]. Endelig kan mikroaspiration af ventrikelindhold være en udløsende årsag til AE-IPF, men hverken behandling med syrepumpehæmmere eller anti refluks-kirurgi er påvist at kunne reducere incidensen [2, 7]. Derimod kan risikoen for AE-IPF reduceres ved behandling af den underliggende IPF med antifibrotisk medicin [8]. Andre forebyggende tiltag er ikke systematisk undersøgt, men kan rettes mod de kendte udløsende årsager.
AE-IPF har kliniske og histopatologiske ligheder med acute respiratory distress syndrome, idet begge kendetegnes af akut respiratorisk forværring, bilaterale matglasforandringer og konsoliderende infiltrater på CT af thorax samt et histopatologisk diffuse alveolar damage-mønster [9].
Det er vigtigt at skelne mellem AE-IPF og den generelle progression af IPF, da patofysiologi, behandling og prognose adskiller sig væsentligt.
Symptomerne ved AE-IPF tiltager over dage til uger med en typisk varighed på under en måned. Der ses forværring af dyspnø (100%), forværring af hoste (ca. 50-80%), ekspektoration (ca. 50-65%) og feber (ca. 25%) [4, 9].
Objektivt ses takypnø og hypoxæmi samt behov for ekstra ilttilskud [4]. Værdien af stetoskopi er begrænset, fordi der ved underliggende IPF kan høres bilateral basal krepitation (såkaldt velcrokrepitation), som kan være svær at skelne fra krepitation ved f.eks. lungeødem eller pneumoni.
Udredning af AE-IPF er beskrevet i Figur 1. Ud over højopløseligheds-CT af lungerne (HRCT), hvormed man kan påvise de typiske radiologiske forandringer, kræves grundig undersøgelse for evt. udløsende faktorer og udelukkelse af anden årsag til den respiratoriske forværring (Tabel 1 og Figur 1).
Billeddiagnostik
Røntgen af thorax og lunge-UL-skanning kan bruges til udelukkelse af differentialdiagnoser til AE-IPF (Tabel 1) som f.eks. pneumothorax eller pleuraeffusion, men undersøgelserne er ikke tilstrækkelige til, at AE-IPF kan bekræftes eller udelukkes.
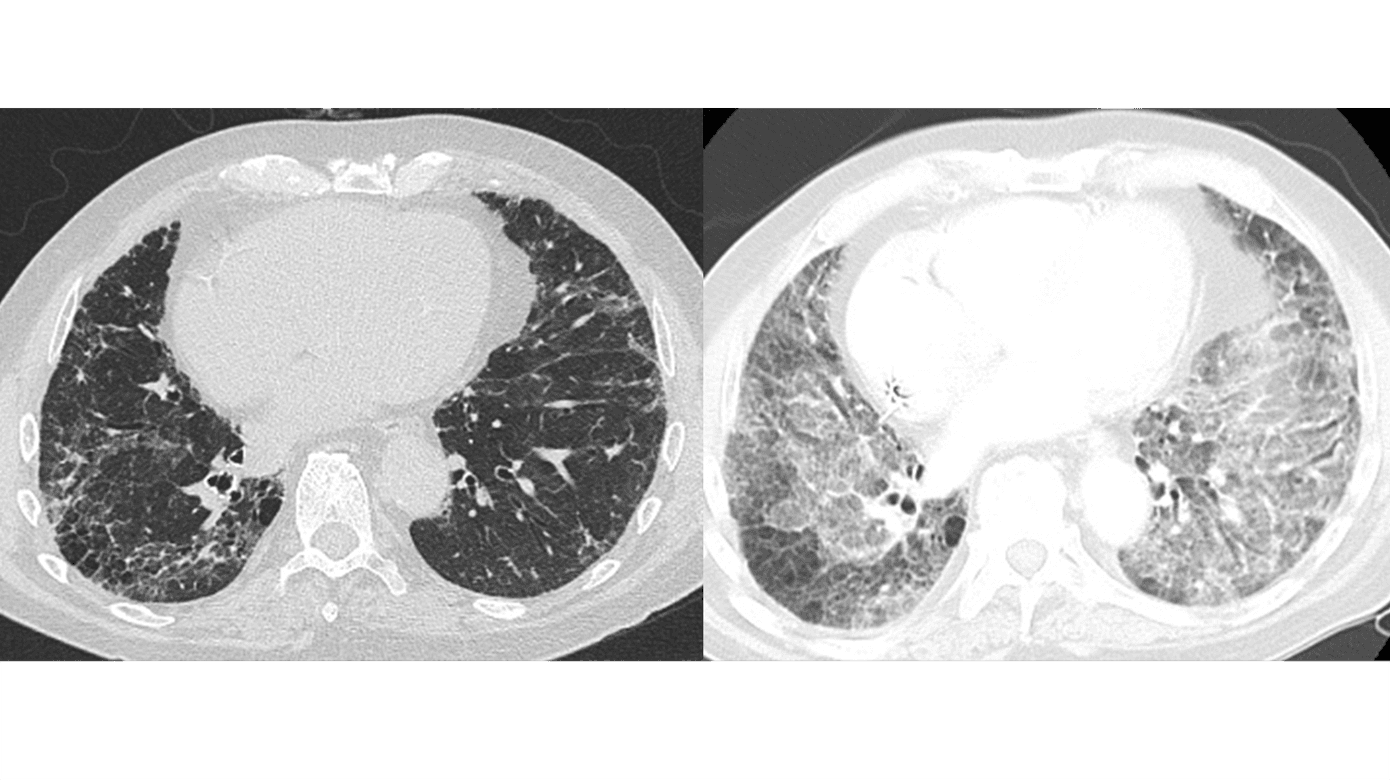
HRCT er nødvendig for at kunne stille diagnosen AE-IPF. Ved AE-IPF ses nyopstået bilateral matglastegning (hyppigst) og/eller konsolidering (sjældnere) oven i et underliggende kendt eller nydiagnosticeret usual interstitial pneumonia (UIP)-mønster (Figur 2) [10]. Disse radiologiske fund indgår i de diagnostiske kriterier for AE-IPF (Tabel 2) [2].
HRCT bør foretages før evt. CT af thorax med i.v. kontrast (alternativt SPECT/CT), som bruges til udelukkelse af lungeemboli [11]. CT af thorax med i.v. kontrast er ikke optimal til vurdering af matglastegning, idet kontrastindgift og den ofte manglende maksimale inspiration vanskeliggør vurdering af lungeparenkymet.
Øvrige undersøgelser
Ingen blodprøver er diagnostiske for AE-IPF, men tages mhp. differentialdiagnostik. Typisk måles der CRP, leukocytter med differentialtælling, procalcitonin, D-dimer, koronarmarkører og evt. pro-BNP. Akutte fasereaktanter er imidlertid ofte forhøjede og kan derfor ikke bruges til sikkert at skelne AE-IPF fra f.eks. infektion eller lungeemboli [4].
Bloddyrkning og nedre luftvejssekret til dyrkning og resistensundersøgelse inkl. undersøgelse for atypiske bakterier samt luftvejsvira tages fra alle patienter. Ofte undersøges også for opportunistisk infektion som f.eks. pneumocyster [9, 11].
Ekkokardiografi er relevant til påvisning af hjertesvigt eller indikation af pulmonal hypertension, som er en hyppig komplikation hos patienter med svær IPF. Bronkoskopi med mikrobiologisk bronkoalveolær lavage (BAL) kan overvejes i udvalgte tilfælde, men der er risiko for forværring af tilstanden, og udbyttet er ofte begrænset [12]. Differentialcelletælling af BAL-væske under en AE-IPF bidrager ikke væsentligt til diagnosen, da både AE-IPF og infektion giver et øget antal neutrofilocytter i BAL-væsken [9, 12]. Lungebiopsi frarådes pga. høj risiko for død [13].
Evidensgrundlaget for behandlingsmulighederne ved AE-IPF er svagt og ofte begrænset til ekspertviden, hvorfor der ses variation i behandlingen mellem forskellige lande [11].
Glukokortikoider og immunosuppressiva
I internationale guidelines anbefales som udgangspunkt glukokortikoider til de fleste patienter med AE-IPF (anbefaling baseret på svag evidens) [11, 14]. Der er ikke gennemført RCT’er, hvor man har undersøgt indikation, dosis eller varighed af behandlingen med glukokortikoider, men i 2023 er der igangsat et RCT, hvor man undersøger effekten af methylprednisolon over for placebo ved AE-IPF [15]. Nyere ekspertholdninger hælder mod lavere kortikosteroiddoser end dem, der hidtil er brugt.
Afhængigt af sværhedsgraden af AE-IPF gives glukokortikoid i form af enten prednisolon 0,5-1 mg/kg dgl. eller methylprednisolon 500-1.000 mg givet i.v. dgl. i tre dage, herefter aftrappet til prednisolon 0,5 mg/kg dgl.
Der foreligger ikke stærk evidens for behandlingsvarighed, men baseret på ekspertviden anbefales ved klinisk respons nedtrapning af glukokortikoiddosis over uger til måneder [11].
Andre immunosuppressiva bliver ikke brugt i behandlingen af AE-IPF. Tillæg af cyclophosphamid til methylprednisolon gav i et RCT øget dødelighed hos patienter med AE-IPF sammenlignet med højdosis methylprednisolon alene (tremånedersmortalitet 45% vs. 31%) [16]. Tillæg af rekombinant humant thrombomodulin til methylprednisolon har, på trods af gode resultater i observationelle undersøgelser, ikke kunnet øge overlevelsen ved AE-IPF i et RCT [17].
Antifibrotisk medicin
Den generelle fibrosedannelse ved IPF kan nedsættes ved fast tabletbehandling med antifibrotiske medikamenter (nintedanib eller pirfenidon), som også er vist at mindske risikoen for AE-IPF [8, 18, 19]. Derimod er effekten af at starte behandling med antifibrotisk medicin under en AE-IPF usikker, og individuel vurdering og drøftelse med et ILS-center anbefales [20]. Antifibrotisk medicin øger ikke risikoen for infektioner, og hvis patienten allerede er i behandling med pirfenidon eller nintedanib, fortsættes denne uændret under en AE-IPF [11].
Øvrig medicinsk behandling
Ekstra ilttilskud er ofte nødvendigt ved AE-IPF, evt. som high-flow ilttilskud. Patienter med IPF kan som regel tåle ilttilskud uden at udvikle hyperkapni, medmindre de samtidig har KOL/emfysem.
Patienter med AE-IPF har øget risiko for dyb venetrombose og lungeemboli og bør derfor gives profylaktisk behandling med lavmolekylært heparin [21].
Infektion kan være en udløsende årsag til en AE-IPF. Derfor gives der som regel antibiotika og evt. anden antimikrobiel behandling på empirisk grundlag, indtil PCR- samt dyrknings- og resistenssvar foreligger [11, 22].
Det foreligger ingen data for behandling af AE-IPF med den inhalerede prostacyclinanalog treprostinil, som muligvis kan bruges i behandlingen af pulmonal hypertension ved fibrotisk ILS [23].
Mekanisk ventilation
Respiratorbehandling kan være relevant i udvalgte tilfælde, især hvis patienterne allerede er på venteliste til lungetransplantation. Beslutningen skal dog holdes op imod en etårsmortalitet på op til 90% hos respiratorkrævende patienter med AE-IPF [14, 24, 25].
Få selekterede patienter med AE-IPF kan komme i betragtning til lungetransplantation, og drøftelse med et transplantationscenter er nødvendig, inden man træffer beslutning om mekanisk ventilation eller ekstrakorporal membranoxygenering.
Noninvasiv ventilation (NIV) er ikke kontraindiceret ved AE-IPF, men hyperkapnisk respirationssvigt ses som regel kun ved samtidig KOL/emfysem [11]. I et mindre studie fandt man, at NIV havde en acceptabel sikkerhedsprofil ved AE-IPF [26].
Palliation
Palliation skal tænkes ind i alle dele af behandlingen af AE-IPF. Ilt, morfin, evt. inhaleret bronkodilaterende medicin og lindring af andre symptomer (smerter, obstipation, depression, refluks etc.) er væsentlige dele af behandlingen.
Beslutning om behandlingsniveau og behandlingsophør kan blive relevant efter inddragelse af patient og pårørende.
Prognosen ved AE-IPF er alvorlig, selv med behandling. De præcise estimater for mortalitet afhænger af den undersøgte population samt den benyttede definition og sværhedsgraden af AE-IPF.
Hospitalmortalitet af AE-IPF trods behandling er i observationelle studier fundet at være op til 50%, og medianoverlevelsen efter eksacerbation er 2-4 mdr. [2, 4, 25]. Dog er der i placebogruppen i et randomiseret studie med patienter med AE-IPF fundet en noget lavere mortalitet, idet man fandt en tremånedersmortalitet på 31% [16]. Overlevere oplever ofte klinisk betydende og irreversibel forværring af lungefunktion, funktionsniveau samt livskvalitet [3, 16]. Overlevere, som ikke allerede er udredt til lungetransplantation, bør vurderes mhp. dette.
Der er beskrevet akutte eksacerbationer for de fleste interstitielle lungesygdomme (AE-ILS), f.eks. reumatoid artritis-associeret ILS, fibrotisk allergisk alveolitis og uklassificerbar fibrotisk ILS, med en estimeret etårsmortalitet på 33-100%, afhængigt af den underliggende ILS [27, 28].
Ofte bruges de samme kriterier for AE-ILS som for AE-IPF (Tabel 2), herunder kriteriet om allerede kendt eller formodet underliggende fibrotisk ILS [27, 29]. Dog vil det underliggende fibrotiske mønster ikke nødvendigvis være et UIP-mønster som ved IPF.
Der foreligger ingen RCT’er, hvori det undersøges, hvordan AE-ILS optimalt håndteres. Der bruges ofte den samme udredning og behandling som for AE-IPF med stort fokus på udelukkelse af differentialdiagnoser samt understøttende og pallierende behandling. Det kan være særlig relevant at undersøge for medicinudløst pneumonitis, hvor seponering af det udløsende agens og behandling med glukokortikoid kan være livreddende, samt infektioner, da patienter med non-IPF fibrotisk ILS hyppigt behandles med immunosuppressiva [28].
Behandlingen af AE-ILS er kompleks, da der ofte er behov for en individualiseret tilgang for de forskellige ILS. F.eks. kan der ved autoimmune ILS med primært inflammatorisk patologi lægges større vægt på immunosuppressiva, og for systemisk sklerodermi-associeret ILS anbefales forsigtighed med høje doser glukokortikoid pga. risiko for udvikling af malign hypertension og renal krise [30]. Patienter med AE-ILS bør derfor konfereres med et af landets højtspecialiserede ILS-centre.
AE-IPF og AE-ILS er akutte forværringer af den underliggende fibrotiske lungesygdom, kendetegnet ved respiratorisk forværring med signifikant ny alveoleskade. Diagnosen stilles ved karakteristiske forandringer på HRCT og grundig udelukkelse af differentialdiagnoser. Behandlingen baseres på glukokortikoid og understøttende behandling, men mortaliteten er høj på trods af behandling.
Korrespondance Saher Burhan Shaker. E-mail: saher.burhan.shaker@regionh.dk
Antaget 13. juni 2023
Publiceret på ugeskriftet.dk 14. august 2023
Interessekonflikter Der er anført potentielle interessekonflikter. Forfatternes ICMJE-formularer er tilgængelige sammen med artiklen på ugeskriftet.dk
Referencer findes i artiklen publiceret på ugeskriftet.dk
Artikelreference Ugeskr Læger 2023;185:V04230261
Nils Hoyer, Elisabeth Bendstrup, Jesper Rømhild Davidsen, Thomas Kromann Lund & Saher Burhan Shaker
Ugeskr Læger 2023;185:V04230261
Acute exacerbation of idiopathic pulmonary fibrosis (AE-IPF) and other fibrotic interstitial lung diseases (AE-ILD) is defined by significant acute respiratory worsening and new widespread alveolar damage. This review summarises the current knowledge of diagnosis and treatment of these events. The diagnosis of AE-IPF and AE-ILD is based on typical HRCT findings of new and bilateral ground glass opacification and/or consolidation, and exclusion of fluid overload or cardiac failure. Treatment relies, despite low quality of evidence, on glucocorticoid in addition to supportive and palliative treatment. Despite treatment, the prognosis is poor, with a median survival of 2-4 months.