Statusartikel
Amyloidose er en sygdom med mange ansigter
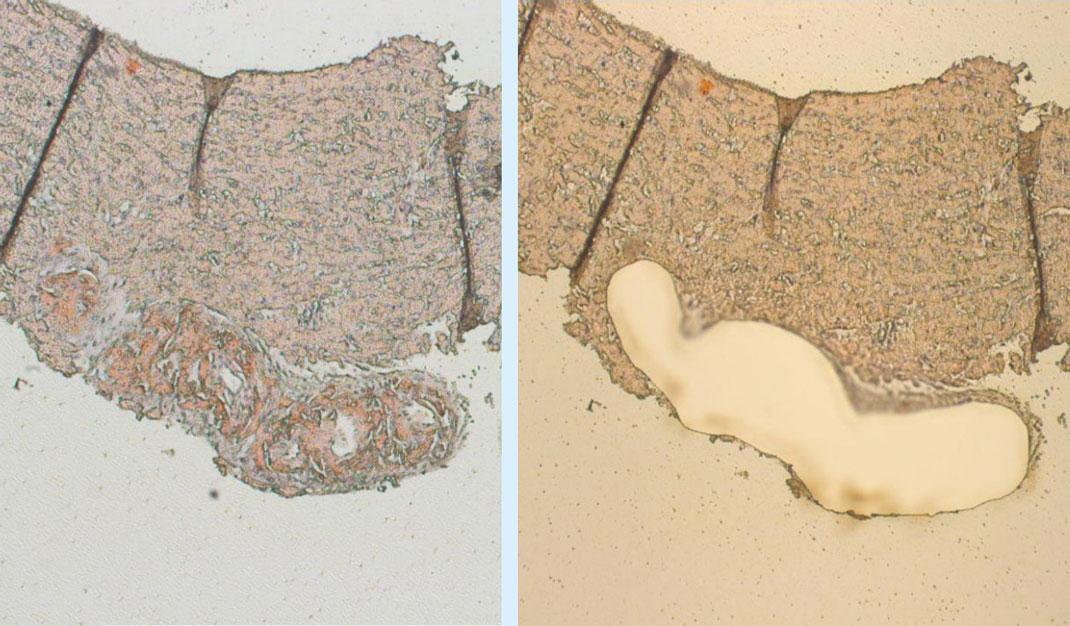
Myokardiebiopsi før og efter laserdissektion af område med amyloid substans.
2. jan. 2023
12 min.
Amyloidose er en alvorlig sygdom, hvis kliniske præsentation kan ligne mange andre sygdomme. Det er en proteinaflejringssygdom, hvor amyloidogene proteiner aggregerer og deponeres ekstracellulært i væv og organer som fibrillære strukturer i en lysmikroskopisk amorf masse, som er positiv i congorød farvning.
Fakta
Amyloidose er en alvorlig proteinaflejringssygdom, hvis heterogene sygdomsbillede besværliggør og forsinker diagnosen.
Hurtig diagnosticering, sikker subtypebestemmelse og tidligt indsat behandling er afgørende for prognose og livskvalitet.
Amyloidose er en sygdom, der kan og skal behandles.
Der er mindst 38 beskrevne amyloidogene proteiner med let immunglobulinkæde-amyloid (AL) og transthyretinamyloid (ATTR) som de to klart mest almindelige former i Danmark (Tabel 1) [1]. Nogle subtyper er arvelige, andre er erhvervede. Behandlingen af de enkelte subtyper er dog meget forskellig, og sikker diagnostisk subklassifikation er derfor helt afgørende.
De afgørende nyheder inden for amyloidose, som berettiger en statusartikel i Ugeskrift for Læger, er, at ny molekylær diagnostik muliggør sikker subtypebestemmelse af amyloidet, kombineret med at nye medicinske behandlinger ved AL- og ATTR-amyloidose har forbedret prognosen markant. Derfor er øget opmærksomhed på sygdommen og tidlig diagnostik afgørende for forbedret prognose og livskvalitet for patienterne. Det store mantra er tidlig awareness og diagnostik. Det heterogene symptombillede og de varierende organmanifestationer resulterer desværre i, at sygdommen ofte overses, og diagnosetidspunktet forsinkes [2-4].
De kliniske manifestationer af amyloidose afhænger af involveret vævstype og organ samt sværhedsgraden af aflejringen [5]. De hyppigst involverede organsystemer er hjerte, nyrer, nervesystem og mave-tarm-kanal, og ofte er mere end et organsystem afficeret [2, 5, 6]. I Tabel 2 ses, hvordan involveringen af de enkelte organer typisk præsenterer sig, og der fremhæves tidlige varslingssymptomer, som bør rette mistanke om amyloidose som mulig differentialdiagnose.
I det følgende beskrives i kort form særlige opmærksomhedspunkter omkring de affødte konsekvenser af amyloidaflejring i de enkelte organer.
Hjerte
Ved kardial involvering ses typisk hypertrofisk hjerte, diastolisk dysfunktion og restriktivt hjertesvigt [7] og i mere fremskredne stadier også systolisk hjertesvigt. Symptomer omfatter dyspnø, træthed og væskeophobning. Hos patienter med hjertesvigt med bevaret uddrivningsfraktion er årsagen amyloidose hos op mod 15%, hvorfor dette fund, særligt hos ældre mænd, bør give mistanke om amyloidose [4]. Atriel arytmi og overledningsforstyrrelser er hyppigt forekommende. Atrieflimren ses hos op til 70% af patienter med kardial ATTR-amyloidose og hos 26% af patienter med kardial AL-amyloidose [2-4], og op mod en tredjedel af patienter med kardial ATTR-amyloidose vil have behov for pacemaker. Faldende blodtryk og ortostatisme er almindeligt, sidstnævnte dog også ofte betinget af påvirkning af det autonome nervesystem [8].
Nyrer
Det typiske, tidlige tegn på nyrepåvirkning er albuminuri på > 500 mg pr. døgn, men ikke alle subtyper giver betydelig proteinuri. AL- og sekundært amyloid A (AA)-amyloidose giver proteinuri på niveau med nefrotisk syndrom, mens leukocytkemotaksin-2 (ALECT2)-amyloidose giver lav til ingen proteinuri, idet aflejringen primært er interstitiel og vaskulær [3]. Mere avanceret klinisk præsentation er nefrotisk syndrom eller en kombination af proteinuri og nedsat glomerulær filtration [2, 9]. ATTR-amyloidose giver typisk ikke renal aflejring, og eventuel nyrepåvirkning ved ATTR-amyloidose skyldes ofte nedsat perfusion grundet nedsat cardiac output og hypotension [3].
Nervesystem
Perifer neuropati starter ofte som distal, symmetrisk neuropati med neuropatiske smerter og nedsat temperatursans, som kan udvikle sig til anæstesi og kraftnedsættelse [3, 6]. Påvirkning af det autonome nervesystem kan medføre en række vage symptomer, men præsenterer sig ofte som ortostatisk hypotension, erektil dysfunktion, nedsat tarmperistaltik, diarré og obstipation [3].
Muskel- og bindevæv
Karpaltunnelsyndrom (KTS) er i høj grad associeret med systemisk AL- og ATTR-amyloidose, og tidligere operation herfor er hyppig i anamnesen, ofte flere år før amyloidosediagnosen stilles. Op mod 50% af patienter med diagnosticeret kardial ATTR-amyloidose har historik med KTS [2, 3]. Omvendt har ca. 15% af patienter, som opereres for KTS-påviseligt amyloid, hyppigst ATTR-amyloidose i bløddelsvævet omkring nervus medianus [10], men majoriteten har ikke anden påviselig organinvolvering. Andre manifestationer af bløddelsaflejringer ved ATTR-amyloidose er senerupturer, f.eks. bicepsseneruptur, og spinalstenose. I fremskredent stadie kan patienter med især AL-amyloidose få spontane periorbitale ekkymoser,»vaskebjørnsøjne«, makroglossi og subkutane aflejringer, f.eks.»skulderpuder« [2, 3].
Amyloidose diagnosticeres ved påvisning af amyloid i biopsi. Ved screening for systemisk amyloidose kan subkutant fedtaspirat bruges som et indledende diagnostisk redskab. I kyndige hænder er undersøgelsen lettilgængelig og forholdsvis smerte- og bivirkningsfri. Sensitivitet ved AL-amyloidose er ca. 70%, men mindre ved ATTR-amyloidose, ca. 40% [11]. Kombineret med knoglemarvsbiopsi stiger sensitiviteten ved AL-amyloidose til over 80% [12], men der vil altså i en del tilfælde være behov for biopsi fra mistænkt afficeret organ [3, 12]. I det følgende gives nogle få vigtige pointer inden for udredning af de enkelte organer.
Kardial udredning
Ekg
Ekg spiller en vigtig rolle ved mistanke om amyloidose. I op til 70% af patienterne kan man se pseudoinfarkter og patologiske Q-takker uden tidligere tilfælde med infarkt [13]. Ved fremskreden sygdom ses low voltage på ekg hos ca. 50% af patienter med AL-amyloidose og hos 25-40% af patienter med ATTR-amyloidose [14]. Lav QRS-amplitude opstår, da den amyloide infiltration virker isolerende på det elektriske signal [14]. Low voltage er et karakteristisk fund, men sensitiviteten er lav, og metoden er derfor ikke egnet som screeningsundersøgelse.
Ekkokardiografi
Ekkokardiografi (EKKO)viser hypertrofi af venstre og eventuelt højre ventrikel, lille ventrikelvolumen og eventuelt fortykkede klapper [14]. Tidligt i forløbet er uddrivningsfraktionen ofte bevaret, men forværres gradvist over tid [14]. Ved almindelig 2D EKKO er sensitiviteten lav, men ved såkaldt global longitudinal strain-analyse, som måler hjertets deformation, ses typisk relativ bevarelse af hjertets apex, et »bull’s eye pattern«, som sjældent ses ved andre former for kardiomyopati [14], og derved øges specificitet og sensitivitet.
Kardial MR-skanning
Kardial MR-skanningmed i.v.-kontrast giver den største sensitivitet og specificitet ved noninvasiv diagnostik af kardial amyloidose og regnes i dag som guldstandard [3, 14]. Den er egnet til at udelukke andre årsager til hypertrofi, evaluere behandlingsrespons og påvise progression af aflejring [14-16]. Ved brug af gadoliniumkontrast opnår man en sensitivitet på 85% og en specificitet på 92% for kardial amyloidose. I kombination med måling af ekstracellulært volumen ved T1-mapping opnås en endnu højere sensitivitet og specificitet [3].
Knoglescintigrafi
Knoglescintigrafihar forbedret muligheden for noninvasiv diagnostik af kardial ATTR-amyloidose. Knoglescintigrafi anvendes i differentialdiagnostikken mellem AL- og ATTR-amyloidose. Ved ATTR-amyloidose ses typisk markant, visuelt traceroptag grad 2-3 i myokardiet (Figur 1), hvorimod der ikke ses optag eller kun ringe optag ved AL-amyloidose. Ved fravær af biokemisk mistanke om plasmacelledyskrasi, dvs. fravær af M-komponent i blod og urin samt fund af normale niveauer af frie kappa- og lambdakæder i serum, har knoglescintigrafi med 99mTc-diphospho-propan-dicarboxylat en sensitivitet på 91% og en specificitet på 100% for kardial ATTR-amyloidose ved traceroptag grad 2-3 [17].
Renal udredning
Der findes ingen noninvasiv procedure til påvisning af amyloid i nyren. Kan amyloid ikke påvises ved fedtaspiration eller knoglemarvsbiopsi, er nyrebiopsi nødvendig for præcis diagnose [9].
Hæmatologisk udredning
Hæmatologiskudredning indebærer som det vigtigste måling af frie lette kappa- og lambdakæder i serum. Denne analyse er ved at blive udrullet nationalt og gjort tilgængelig for praktiserende læger og andre kolleger uden for hæmatologien. Herudover måles M-komponent i serum og urin. Ved knoglemarvsundersøgelse med såvel morfologisk som flowcytometrisk undersøgelse kan det afgøres, om der er plasmacelleklon eller anden B-celle-proliferativ sygdom samt amyloid aflejring i marven [3].
Subtypebestemmelse
Ved påvist amyloid aflejring er næste skridt at sikre præcis bestemmelse af det amyloide protein. Guldstandard for subtypebestemmelse er i dag immunelektronmikroskopi (IEM) og massespektrometri (MS). Kombinationen af disse to undersøgelsesmetoder har en sensitivitet og en specificitet på næsten 100% [18]. I Danmark foretages MS og IEM kun ved Odense Amyloidose Center på Odense Universitetshospital.
Immunelektronmikroskopi
IEM kan visualisere de amyloide fibriller vha. elektronmikroskopi. Ved immunfarvning med guldmarkerede antistoffer kan subtypen bestemmes med en sensitivitet på 91,6% [18]. Som standardmetode bruges kappa-, lambda- og transthyretinantistoffer, der kan suppleres med anti-AA.
Massespektrometri
MS kan detektere amyloide subtypeproteiner i amyloid substans. Congorødpositive områder fridissekeres med laserdissektionsmikroskopi (Figur 2) og trypsindegraderes til peptider, som sekventeres ved tandem-MS. Sekvensinformationen anvendes så til identifikation af det amyloidogene protein ved bioinformatisk analyse. Ud over den specifikke subtype kan MS registrere amyloide signaturproteiner, hvilket fungerer som positiv, intern kontrol [18-20]. Subtypebestemmelse ved MS er forbundet med høj sensitivitet og specificitet på henholdsvis 88,8% og 96% [21].
Overordnet mål
Det overordnede behandlingsmål retter sig mod at nedsætte koncentrationen af amyloid precursor protein for at forhindre yderligere aflejringer [3]. Der findes endnu ikke godkendte behandlingsmuligheder til opløsning af amyloide aflejringer, men flere behandlingsmodeller er på vej. Hurtig diagnostik og initiering af korrekt behandling er afgørende for prognose og livskvalitet hos patienterne [3, 4].
AL-amyloidose
Behandling af AL-amyloidose retter sig mod den underliggende klonale plasmacelledyskrasi og sigter mod at reducere mængden af cirkulerende amyloidogene frie lette kæder [22]. Primærbehandling i dag er en kombination af bortezomib, cyclophosphamid og dexamethason [8, 23]. Tillæg af daratumumab, et monoklonalt antistof rettet mod plasmacelleoverfladeantigenet CD38, har vist forbedret effekt. Kombinationen er godkendt af Det Europæiske Lægemiddelagentur (EMA), men er endnu ikke godkendt af Medicinrådet i Danmark [24]. Udvalgte, yngre patienter kandiderer til højdosiskemoterapi med stamcellestøtte. Præliminære studier har vist, at monoklonale antistoffer rettet mod AL-aflejringer måske kan fremme opløsning af disse [25], og studier heraf pågår.
Transthyretinamyloidose
Der skelnes mellem vildtype-ATTR (ATTRwt)- og hereditær ATTR (ATTRv)-amyloidose [1]. Levertransplantation har været førstelinjebehandling ved ATTRv-amyloidose, da precursorproteinet dannes i leveren [26]. I dag findes medicinske behandlingsmuligheder, og patisiran og inotersen er godkendt til behandling af ATTRv-amyloidose med polyneuropati grad 1 og grad 2. Begge midler er små oligonukleotid-RNA-interfererende stoffer, som specifikt hæmmer syntesen af transthyretin i levercellerne [27, 28]. Forsøg ved ATTRwt-amyloidose pågår. Tafamidis er et antiaggregatorisk virkende lægemiddel, som stabiliserer den tetramere struktur af transthyretin og derved modvirker misfoldning og fibrildannelse, og lægemidlet har effekt ved både ATTRv- og ATTRwt-amyloidose [29]. I Danmark anbefaler Medicinrådet alene tafamidis til visse undergrupper med ATTRv-amyloidose.
Andre typer amyloidose
Behandlingen af AA-amyloidose retter sig mod den underliggende inflammatoriske tilstand [6].Der findes ingen behandlingsguidelines for ALECT2-amyloidose, men nyretransplantation har i nogle tilfælde været anvendt [9]. Lokal amyloidose kan findes i hud, øjne, luftveje og urogenitalier. Behandlingen er typisk resektion af afficeret område [30].
Amyloidose er en alvorlig proteinaflejringssygdom. Der er i dag kendskab til mindst 38 forskellige proteiner med amyloiddannende egenskaber, hvor AL og ATTR er langt de hyppigst forekommende. Det heterogene symptombillede og den sjældne forekomst resulterer desværre ofte i, at sygdommen overses, eller diagnosen forsinkes. Sygdomsopmærksomhed, hurtig diagnostik, sikker subtypebestemmelse efterfulgt af hurtigt indsat relevant behandling er afgørende for prognose og livskvalitet. I de seneste år er der sket store fremskridt inden for diagnostik og behandling.
Korrespondance Niels Abildgaard. E-mail: niels.abildgaard@rsyd.dk
Antaget 15. november 2022
Publiceret på ugeskriftet.dk 2. januar 2023
Interessekonflikter ingen. Forfatternes ICMJE-formularer er tilgængelige sammen med artiklen på ugeskriftet.dk
Artikelreference Ugeskr Læger 2023;185:V08220479
Magne B. L. Spanggaard, Charlotte T. Hansen, Michael Maiborg, Aleksandra M. Rojek, Sys Vestergaard, Hans Christian Beck, Hanne E. H. Møller & Niels Abildgaard
Ugeskr Læger 2023;185:V08220479
Amyloidosis is a severe disease caused by protein misfolding and deposition in tissues and organs. Thirty-eight different proteins are known to be amyloidogenic. Amyloidosis is categorized into inherited or acquired, and systemic or localized. Light‐chain (AL)- and transthyretin (ATTR) amyloidosis are the two most common subtypes. Awareness, early diagnosis, accurate subtyping and relevant treatment are crucial for the management. Novel therapies of systemic AL and ATTR amyloidosis have considerably improved outcome and survival. The aim of this review is to increase awareness and knowledge on diagnosing amyloidosis.
Benson MD, Buxbaum JN, Eisenberg DS et al. Amyloid nomenclature 2020: update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2020;27(4):217-222. doi: 10.1080/13506129.2020.1835263.
Vaxman I, Gertz M. When to suspect a diagnosis of amyloidosis. Acta Haematol. 2020;143(4):304-311. doi: 10.1159/000506617.
Law S, Fontana M, Gillmore JD. Advances in diagnosis and treatment of cardiac and renal amyloidosis. Cardiol Clin. 2021;39(3):389-402. doi: 10.1016/j.ccl.2021.04.010.
Nativi-Nicolau JN, Karam C, Khella S, Maurer MS. Screening for ATTR amyloidosis in the clinic: overlapping disorders, misdiagnosis, and multiorgan awareness. Heart Fail Rev. 2022;27(3):785-793. doi:10.1007/s10741-021-10080-2.
Thomas VE, Smith J, Benson MD, Dasgupta NR. Amyloidosis: diagnosis and new therapies for a misunderstood and misdiagnosed disease. Neurodegener Dis Manag. 2019;9(6):289-299. doi: 10.2217/nmt-2019-0020.
Muchtar E, Dispenzieri A, Magen H et al. Systemic amyloidosis from A (AA) to T (ATTR): a review. J Intern Med. 2021;289(3):268-292. doi: 10.1111/joim.13169.
Bhogal S, Ladia V, Sitwala P et al. Cardiac amyloidosis: an updated review with emphasis on diagnosis and future directions. Curr Probl Cardiol. 2018;43(1):10-34. doi: 10.1016/j.cpcardiol.2017.04.003.
Ihne S, Morbach C, Sommer C et al. Amyloidosis - the diagnosis and treatment of an underdiagnosed disease. Deutsch Ärztebl Int. 2020;117(10):159-166. doi: 10.3238/arztebl.2020.0159.
Gupta N, Kaur H, Wajid S. Renal amyloidosis: an update on diagnosis and pathogenesis. Protoplasma. 2020;257(5):1259-1276. doi: 10.1007/s00709-020-01513-0.
Sekijima Y, Uchiyama S, Tojo K et al. High prevalence of wild-type transthyretin deposition in patients with idiopathic carpal tunnel syndrome: a common cause of carpal tunnel syndrome in the elderly. Hum Pathol. 2011;42(11):1785-91. doi: 10.1016/j.humpath.2011.03.004.
Hansen CT, Møller HEH, Rojek AM et al. Combined subcutaneous fat aspirate and skin tru-cut biopsy for amyloid screening in patients with suspected systemic amyloidosis. Molecules. 2021;26(12):3649. doi: 10.3390/molecules26123649.
Cohen OC, Sharpley F, Gilbertson JA et al. The value of screening biopsies in light‐chain (AL) and transthyretin (ATTR) amyloidosis. Eur J Haematol. 2020;105(3):352-356. doi: 10.1111/ejh.13458.
Rubin J, Maurer MS. Cardiac amyloidosis: overlooked, underappreciated, and treatable. Annu Rev Med. 2020;71(1):203-219. doi: 10.1146/annurev-med-052918-020140.
Martinez-Naharro A, Baksi AJ, Hawkins PN, Fontana M. Diagnostic imaging of cardiac amyloidosis. Nat Rev Cardiol. 2020;17(7):413-426. doi: 10.1038/s41569-020-0334-7.
Maceira AM, Joshi J, Prasad SK et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):186-93. doi: 10.1161/01.CIR.0000152819.97857.9D.
Fontana M, Chung R, Hawkins PN, Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20(2):133-44. doi: 10.1007/s10741-014-9470-7.
Gillmore JD, Maurer MS, Falk RH et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404-12. doi: 10.1161/circulationaha.116.021612.
Abildgaard N, Rojek AM, Møller HEH et al. Immunoelectron microscopy and mass spectrometry for classification of amyloid deposits. Amyloid. 2020;27(1):59-66. doi: 10.1080/13506129.2019.1688289.
Palstrøm NB, Rojek AM, Møller HEH et al. Classification of amyloidosis by model-assisted mass spectrometry-based proteomics. Int J Mol Sci. 2021;23(1):319. doi: 10.3390/ijms23010319.
Dogan A. Amyloidosis: insights from proteomics. Annu Rev Pathol. 2017;12(1):277-304. doi: 10.1146/annurev-pathol-052016-100200.
Vrana JA, Theis JD, Dasari S et al. Clinical diagnosis and typing of systemic amyloidosis in subcutaneous fat aspirates by mass spectrometry-based proteomics. Haematologica. 2014;99(7):1239-47. doi: 10.3324/haematol.2013.102764.
Gertz MA, Dispenzieri A. Systemic amyloidosis recognition, prognosis, and therapy. JAMA. 2020;324(1):79-89. doi: 10.1001/jama.2020.5493.
Ryšavá R. AL amyloidosis: advances in diagnostics and treatment. Nephrol Dial Transplant. 2019;34(9):1460-1466. doi: 10.1093/ndt/gfy291.
Kastritis E, Palladini G, Minnema MC et al. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. 2021;385(1):46-58. doi: 10.1056/nejmoa2028631.
Chandrashekar P, Desai AK, Trachtenberg BH. Targeted treatments of AL and ATTR amyloidosis. Heart Fail Rev. 2022;27(5):1587-1603. doi: 10.1007/s10741-021-10180-z.
Gertz MA, Benson MD, Dyck PJ et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-2466. doi: 10.1016/j.jacc.2015.09.075.
Adams D, Gonzalez-Duarte A, O’Riordan WD et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11-21. doi: 10.1056/nejmoa1716153.
Gertz MA, Scheinberg M, Waddington-Cruz M et al. Inotersen for the treatment of adults with polyneuropathy caused by hereditary transthyretin-mediated amyloidosis. Expert Rev Clin Pharmacol. 2019;12(8):701-711. doi: 10.1080/17512433.2019.1635008.
Rapezzi C, Elliott P, Damy T et al. Efficacy of tafamidis in patients with hereditary and wild-type transthyretin amyloid cardiomyopathy: further analyses from ATTR-ACT. JACC Heart Fail. 2021;9(2):115-123. doi: 10.1016/j.jchf.2020.09.011.
Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39(2):323-45. doi: 10.1016/j.rdc.2013.02.012.