Kasuistik
Faldtendens som debut af Creutzfeldt-Jakobs sygdom
20. feb. 2023
5 min.
Creutzfeldt-Jakobs sygdom er den hyppigste prionsygdom hos mennesker med seks nye tilfælde pr. år i Danmark. Prionerne er smitsomme proteinkomplekser, som erstatter de raske prioner med en misfoldet ikkenedbrydelig isoform, der ophobes i hjernen og medfører celledød.
Der skelnes mellem sporadisk, variant af (erhvervet) og genetisk Creutzfeldt-Jakobs sygdom. Sporadisk sygdom opstår tilfældigt med en gennemsnitsalder på 62 år. Ved variantsygdom er gennemsnitsalderen 29 år, og sygdommen blev kendt som bovin spongiform encefalopati eller kogalskab, da man havde mistanke om smitte fra kontamineret oksekød i England i 1996 [1]. Ved mistanke om genetisk sygdom sendes en blodprøve til analyse for dominant arvelige mutationer i priongenet PRNP.
Creutzfeldt-Jakobs sygdom rammer mænd og kvinder ligeligt. Symptomerne er hurtigt udviklede kognitive og adfærdsforstyrrelser, myoklonus (f.eks. startle respons), pyramidale symptomer (f.eks. hyperrefleksi og Babinski), ekstrapyramidale symptomer (f.eks. langsomhed og rigiditet) samt lillehjernesymptomer med gangbesvær. Real-time quaking-induced conversion (RT-QuIC) test på cerebrospinalvæsken giver en sensitivitet på 92% og en specificitet på 99% for Creutzfeldt-Jakobs sygdom, og den udføres på Patologisk Afdeling, Rigshospitalet [2]. MR-skanning af hovedet giver høj sensitivitet på 83-93% og specificitet på 87-95% for sygdommen [3]. Andre fund, som kan understøtte diagnosen, er elektroencefalografi med bi- eller trifasiske periodiske sharp wave-komplekser, hvilket giver høj specificitet, men lav sensitivitet, samt måling af koncentrationen af protein 14-3-3, som har høj sensitivitet, men lav specificitet, og tau-protein er ofte bedre i diagnostikken end protein 14-3-3 [4]. Der findes ingen behandling, og døden indtræder oftest inden for et år. Differentialdiagnoser inkluderer andre former for demens, paraneoplastiske tilstande, autoimmun encefalitis, psykiatriske lidelse eller metaboliske forstyrrelser (f.eks. hypoglykæmi og hjertestop).
En 77-årig kvinde, kendt med radikalopereret mammacancer, men ellers rask, debuterede med gradvist tiltagende gangbesvær og faldtendens, hvor hun over nogle måneder begyndte at falde flere gange dagligt. Hun mente ikke selv, at hun havde problemer, og ønskede sig konsekvent udskrevet under faldudredningen i geriatrisk regi. Desuden ønskede hun at fortsætte bilkørsel, selvom hun ikke kunne gå uden støtte. Hendes tale og skrift havde desuden ændret sig (hun var pensioneret afdelingsleder som bibliotekar og uddannet fra Niels Brock). Hun var ifølge sin datter blevet mere vredladen, men havde ingen hukommelsesbesvær eller øvrige klager.
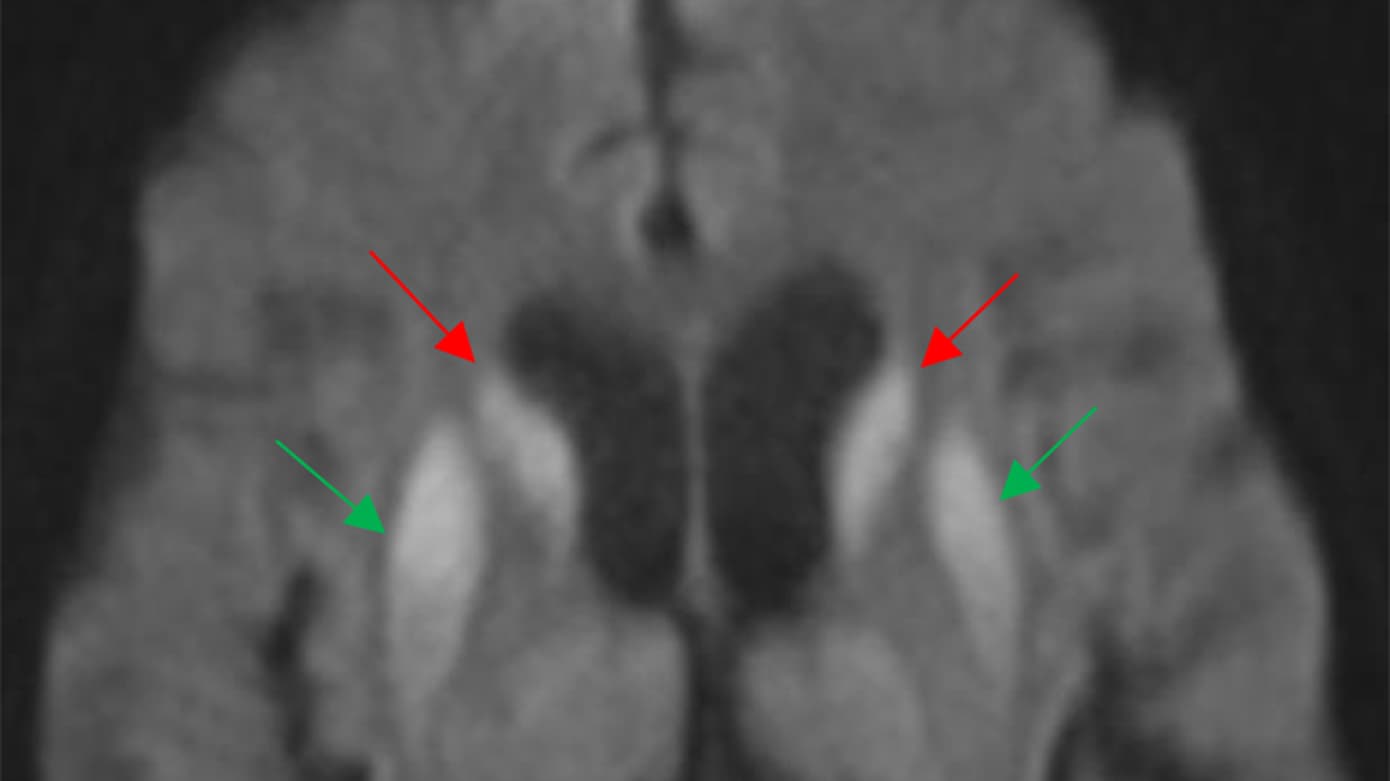
I forbindelse med faldudredning udførtes MR-skanning af hjernen, som viste isolerede hyperintense signaler med kraftig diffusionsrestriktion svarende til nucleus caudatus og nucleus lentiformis (basalganglierne) på begge sider (Figur 1). Hun blev derfor overflyttet til Neurologisk Afdeling, hvor man objektivt fandt hende uden sygdomsindsigt og med tegn fra lillehjernen, herunder opbrudte øjenbevægelser, ataksi ved finger-næse-finger-forsøg samt udtalt trunkal instabilitet som årsager til gangbesværet.
Hun klarede kognitive test rimeligt godt (Mini-Mental State Examination 28/30 og Addenbrooke’s Cognitive Examination 80/100) bortset fra udtalte visuospatiale udfordringer på urskivetesten.
Blodprøver og elektroencefalografi var upåfaldende. Lumbalpunktur, inklusive celletal, protein- samt paraneoplastiske og autoimmun encefalitis-prøver, var normal fraset følgende: forhøjet tau-protein, let nedsat betaamyloid og forhøjet neurofilament light chain. Hendes RT-QuIC test på rygmarvsvæske var positiv.
Patienten havde hurtigt udviklet visuospatiale udfordringer med manglende sygdomsindsigt samt cerebellare symptomer, og hendes MR-skanning viste klassiske tegn på Creutzfeldt-Jakobs sygdom. Da hendes RT-QuIC test var positiv, opfyldte hun kriterierne for mulig sporadisk Creutzfeldt-Jakobs sygdom.
Diagnosen mulig Creutzfeldt-Jakobs sygdom stilles enten ved neuropsykiatriske symptomer og positiv RT-QuIC test eller ved progressiv demens og mindst to af følgende kliniske tegn: 1) myoklonus, 2) visuelle eller cerebellare udfald, 3) pyramidale eller ekstrapyramidale symptomer og 4) akinetisk mutisme, dvs. stumhed og fravær af bevægelser med undtagelse af bevarede følgebevægelser med øjnene. En sikker diagnose stilles ved biopsi, som viser spongiforme forandringer, reaktiv gliose uden inflammation og ophobning af abnormt prionproteinkompleks.
Alle patienter med mulig Creutzfeldt-Jakobs sygdom bør obduceres. Dette gøres dels for at stille en sikker diagnose, men også for at monitorere muligt erhvervede tilfælde. På hjernebiopsi er det muligt at skelne mellem sporadisk og variantsygdom, fordi det patologiske prionprotein er mere stereotypt ved variantsygdom [5].
Sundhedspersonale er ikke i øget risiko for Creutzfeldt-Jakobs sygdom efter kontakt med patienterne, men der skal tages særlige forholdsregler ved håndtering af patienternes cerebrospinalvæske/hjernevæv.
Korrespondance Magnus Spangsberg Boesen. E-mail:
magnus.spangsberg.boesen@regionh.dk
Antaget 12. januar 2023
Publiceret på ugeskriftet.dk 20. februar 2023
Interessekonflikter ingen. Forfatternes ICMJE-formularer er tilgængelige sammen med artiklen på ugeskriftet.dk
Referencer findes i artiklen publiceret på ugeskriftet.dk
Artikelreference Ugeskr Læger 2023;185:V11220697
Patrick Vincent Marloth, Martin Riis Ladefoged & Magnus Spangsberg Boesen
Ugeskr Læger 2023;185:V11220697
Creutzfeldt-Jakob disease is the most common prion disease in humans. Neuropsychiatric symptoms are common, and objective findings are myoclonus, pyramidal and extrapyramidal including cerebellar dysfunction. This is a case report of a 77-year-old woman with gradual onset of repeated falls due to cerebellar dysfunction. She had severe visuospatial difficulties, and she was unaware of her problems. Her MRI showed increased diffusion restriction in the caudate and lentiform nuclei. Her cerebrospinal fluid real-time quaking-induced conversion test was positive, fulfilling criteria for probable sporadic Creutzfeldt-Jakob disease.