Statusartikel
Manifestationer af hepatiske arteriovenøse malformationer ved hereditær hæmoragisk telangiektasi
27. mar. 2023
12 min.
Hovedbudskaber
Hereditær hæmoragisk telangiektasi (HHT), også kendt som Osler-Weber-Rendus sygdom, er en autosomal dominant arvelig sygdom, hvor der dannes arteriovenøse malformationer (AVM) i flere organsystemer. HHT har en prævalens på 1/6.500 i Danmark [1]. Sygdommen diagnosticeres klinisk i henhold til Curaçao-kriterierne: 1) spontant opstående og gentagne tilfælde af epistaxis, 2) flere mukokutane telangiektasier på typiske steder, herunder næseslimhinde, ansigt, læber, mundslimhinde og fingre, 3) AVM i indre organer og 4) familiehistorik med førstegradsslægtning med HHT [1]. Hvis en patient har tre eller flere af ovenstående symptomer, er diagnosen sikker, ved to af ovenstående symptomer er diagnosen mulig, og ved ingen eller et af symptomerne er diagnosen usandsynlig. Diagnosen kan hos omkring 90% bekræftes med gentest [2].
HHT inddeles i HHT type 1 (HHT1) og HHT type 2 (HHT2) samt juvenil polypose (JP)-HHT. Undertyperne er defineret ved den specifikke genmutation, med mutation i henholdsvis endoglin-genet (HHT1), activin A receptor type II-like 1-genet (HHT2) og SMAD4-genet (JP-HHT) [2]. Uanset HHT-subtype er det fælles for mutationerne, at de spiller vigtige roller i angiogenesen [3]. Påvirkning af angiogenesen kan forårsage udvikling af mindre telangiektasier eller større AVM. Telangiektasier og AVM dannes ved, at der sker en dilatation i den postkapillære venole, hvorved blodet passerer direkte fra arterioler til venoler [4]. Hyppigst findes AVM i hud, slimhinder, gastrointestinalkanal, lever, lunger og hjerne [5].
På nuværende tidspunkt kan patienter med HHT i hele Danmark følges på HHT-Centret, der er tilknyttet Øre-Næse-Hals-Afdelingen, Odense Universitetshospital. Kontrollen af patienterne tager udgangspunkt i internationale guidelines fra 2011 og 2020 [5, 6]. Aktuelt screenes og behandles patienterne for pulmonale AVM, de behandles for recidiverende epistaxis, og hvis relevant udredes de for henholdsvis cerebrale AVM og hepatiske AVM (HAVM) i tæt samarbejde med specialespecifikke afdelinger.
Initialt fandt man, at prævalensen af HAVM ved HHT var lav, men med betydelig variation (8-31%), dog har flere nyere studier vist en væsentligt højere prævalens (44-78%) [7-9]. Det anslås, at der i Danmark er 400-650 patienter med HHT og HAVM. Studierne viser ligeledes, at leverinvolveringen hyppigst rammer kvinder over 48 år med HHT2 [7-9]. Ydermere er det vist, at omkring 8% af patienter med HHT har symptomer som følge af HAVM, og et enkelt studie har vist, at antallet af HAVM øges over tid [10]. Symptomerne er som regel relateret til hjertesvigt og består primært af åndenød og perifere ødemer, undertiden ses der dog leverspecifikke symptomer såsom gulsot og ascites.
Det er vigtigt at kende leverens særprægede vaskulære system for at kunne forstå den komplekse patofysiologi hos patienter, som er symptomatiske for HHT og HAVM. Leveren modtager blod fra henholdsvis a. hepatica og v. porta. Højtryks, oxygeneret blod fra a. hepatica, der står for 20-25% af blodforsyningen til leveren, møder lavtryks, energiholdigt blod fra v. porta i de hepatiske sinusoider. Herfra løber blodet igennem leverparenkymet, inden det forlader leveren igennem v. hepatica til v. cava inferior [11].
I 2006 definerede Cho et al, at der anatomisk kunne defineres tre typer af shunts: Type 1 er en direkte adgang mellem en arterie og en eller flere vener, type 2 har flere arterier, som samles i en vene, og i type 3 samles multiple arterier i flere vener, uden at blodforsyningen kommer i forbindelse med de omkringliggende kapillærer. Disse strukturer kaldes en nidus. Der er lav karmodstand gennem hver nidus i modsætning til kapillærerne, og dette fører til et øget flow igennem nidus med en kaskadereaktion til følge. Samlet set kan dette blandt andet føre til nedsat fylde i parallelle kapillærer, og derved opstår der et arterielt »steal«-fænomen førende til lokal iskæmi [12, 13]. De hepatiske AVM er oftest en type 3-karmalformation.
Specifikt i relation til leveren forekommer der anatomisk to slags intrahepatiske shunts og en ekstrahepatisk shunt, som er dannet af ovenstående nidusstrukturer. Intrahepatisk ses: 1) arteriovenøs shunt (a. hepatica til v. hepatica), 2) arterioportal shunt (a. hepatica til v. porta), og ekstrahepatisk ses portovenøs shunt (v. porta til v. hepatica eller v. cava inferior) som afbildet i Figur 1 [14-16].
På trods af, at patienter kan besidde flere af shunttyperne, er der funktionelt oftest kun én af de ovenstående, der er dominerende. Arteriovenøse shunts er den dominerende shunttype, hvor arterielt blod shuntes direkte til det venøse kredsløb, hvilket giver anledning til højtryks blodgennemstrømning til højre side af hjertet. Dette resulterer i højt preload medførende dilatation af den højre hjertehalvdel og fører til højresidigt high-output hjertesvigt (HOCF). Symptomatisk præsenterer patienterne typiske hjertesvigtsymptomer i form af åndenød og deklive ødemer. Denne shunttype har vist at øge risikoen for udvikling af atrieflimren [10, 16, 17]. Den arterieportale shunt, som er næsthyppigst, bevirker, at højtryksblod passerer fra a. hepatica til v. porta. Dette fører til portal hypertension og giver ofte anledning til ascites. Ved portovenøs shuntning er der samtidig en øget risiko for portosystemisk encefalopati, om end det er en sjælden manifestation [10, 16, 17].
Fælles for alle shunts er, at der forekommer en manglende parenkymatøs blodgennemstrømmning i leveren. Dette fænomen kan medføre iskæmisk påvirkning af leverparenkymet og derved føre til remodulering af vævet. Dette øger risikoen for dannelsen af fibrøst væv, som i litteraturen kaldes pseudocirrose [15, 17]. Ligeledes er der højere risiko for dannelse af fokal nodulær hyperplasi (FNH) og nodulær regenerativ hyperplasi (NRH), som klinisk kan være svær at skelne fra hepatocellulært karcinom (HCC) [5, 10, 18]. I tilfælde af arterielt »steal«-fænomen kan ses biliær iskæmi og biliær nekrose betinget af hypoperfusion i det peribiliære plexus samt mesenterial iskæmi [9, 15, 17]. Biliær iskæmi og biliær nekrose vil kunne danne biliær striktur, som giver symptomer i form af smerter under højre kurvatur, ikterus og paraklinisk kolestatisk leverpåvirkning [16, 17]. Dette er en alvorlig komplikation, som ofte kan medføre kolangitis og i nogle tilfælde blødning fra galdevejene. Endoskopisk terapi kan oftest ikke løse problemet hos disse patienter. Levertransplantation (LTX) kan i disse tilfælde være eneste behandlingsmulighed.
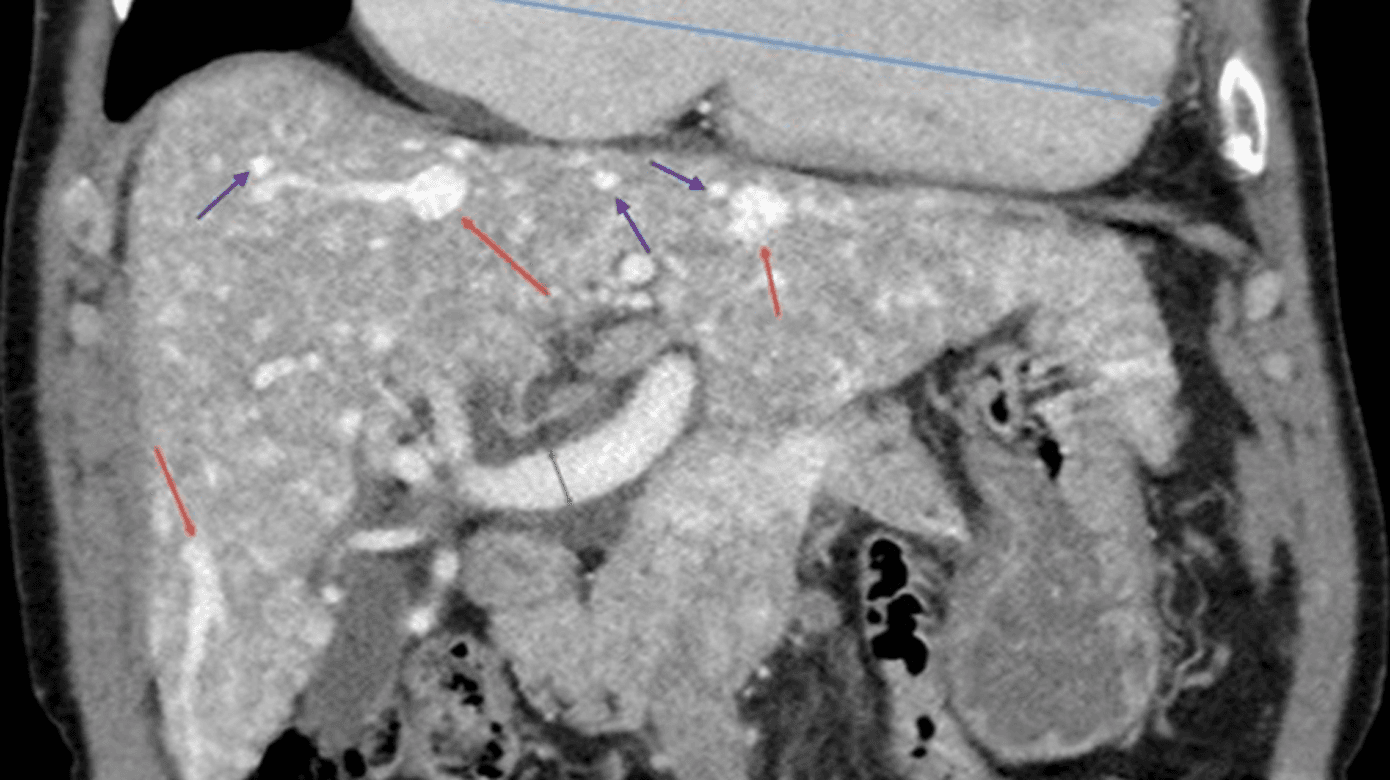
I 2020 udarbejdede Faughnan et al en international guideline, som beskriver diagnosticering, håndtering og behandling af HHT [5]. Den relativt sjældne forekomst af HHT vanskeliggør dog en standardisering, og screeningsprogrammer såvel som behandling beror som oftest på den enkeltes klinikers erfaringer. Det anbefales at screene for HAVM i patienter med HHT, om end der ikke er klare retningslinjer for hyppigheden af screeningen. Flere studier viser, at UL-skanning med Dopplerskanning er ligeværdigt med CT af abdomen med kontrast eller MR-skanning af leveren [8, 16]. I Danmark screenes patienter med HHT med trefaset CT (Figur 2) af leveren aktuelt som 40-årige, og senere i tilfælde af symptomer. Patienter med påviste HAVM, hvor der er symptomer på kardial belastning, tilbydes ekkokardiografi og evt. højresidig hjertekateterisation. Ved fund af fokale forandringer som NRH, FNH og HCC må man benytte sig af MR-skanning til differentialdiagnostik. Der er øget blødningsrisiko ved bioptering, hvilket derfor er relativt kontraindiceret [5, 8, 15].
Der er ingen større randomiserede undersøgelser, der belyser behandling af HAVM. Flere mere eller mindre invasive behandlingsmodaliteter har været afprøvet, herunder embolisering og kirurgisk ligering af HAVM. Begge behandlingsmodaliteter har vist sig at have kortvarig effekt, HAVM regenereres, og symptomerne kommer igen. Ved embolisering har der været beskrevet en del komplikationer i form af iskæmisk kolangitis, iskæmisk kolecystitis, hepatisk nekrose og død [5, 8]. Embolisering og kirurgisk ligering er tidligere blevet anbefalet som palliativ behandling, mens behandlingen nu primært er medicinsk og stiler mod at lindre symptomatisk HAVM-HHT [5].
Aktuelt skelner man mellem symptomatisk behandling og kurativ behandling. Den internationale guideline fra 2020 anbefaler, at behandling af HAVM skal iværksættes ved symptomatiske HAVM. Behandlingen besluttes på baggrund af shunttypen hos den enkelte patient. Hos patienter, der primært har HOCF og pulmonal hypertension, tilbydes medicinsk behandling som ved andre årsager til højresidigt hjertesvigt, diuretika og saltrestriktion [5, 10, 15]. Der er tidligere forsøgt behandling med angiotensinkonverterende enzym-hæmmere og betablokkere, men disse modaliteter har vist sig mangelfulde ved HOCF [19].
Senest er man begyndt at behandle med et anti-vaskulær endotelial vækstfaktor (VEGF)-præparat, bevacizumab (BZB). Der er hos patienter med HHT påvist forhøjede niveauer af VEGF og transforming growth factor-beta, hvilket kan forklare en dysfunktionel angiogenese [20]. BZB benyttes blandt andet til behandling af forskellige cancersygdomme [21]. I flere nyere, ikkerandomiserede studier har man antydet, at BZB-behandling både kan nedsætte blødningstendensen hos patienter med HHT og nedsætte high-output hjertesvigt samt kolangitis betinget af HAVM [8, 22-25]. BZB-behandling har, i små ukontrollerede studier, vist sig at mindske det kardiale output og kliniske symptomer hos 80% af patienter med HOCF [5]. Det er vigtigt at være opmærksom på bivirkningsprofilen af BZB-behandling. De typiske bivirkninger er arteriel hypertension, svær blødning, tromboemboliske episoder, nedsat sårheling, neutropeni og nefrotisk syndrom [21]. Patienter, der har effekt af BZB-behandling, oplever recidiv af symptomerne ved ophør med behandlingen.
Den eneste kurative metode til behandling af HAVM er LTX. Den kliniske erfaring er, at prognosen for patienter med HHT, HAVM og hjertesvigt er dårlig [26]. I 2014 påviste Singh et al, at femårsoverlevelsen hos patienter med HHT og symptomatisk HAVM var 70% [27]. Ydermere viste Reddy et al, at patienter med HOCF forårsaget af shunts af alle årsager havde en femårsoverlevelse på 41% [28]. Lerut et al lavede i 2006 en opgørelse af LTX’er udført i 1985-2003 ved HHT. De påviste, at 17% af patienterne med LTX døde under transplantationen eller i den første uge efter, medførende en tiårsoverlevelse på 83% [29]. Dupuis-Girod et al har opgjort LTX’er udført i Lyon i perioden 1993-2007, og dette studie viser en femårsoverlevelse på 92% [30]. Studiet udførte tillige en livskvalitetsspørgeskemaundersøgelse 3-15 år efter transplantation, og dette spørgeskema viser, at alle de levertransplanterede patienter havde oplevet øget livskvalitet efter transplantationen [30].
HHT en sjælden, arvelig sygdom, hvor genmutationer påvirker angiogenesen. HAVM er en hyppig problemstilling, oftest med få kliniske symptomer. Hos enkelte patienter kan HAVM være medvirkende faktor til tidlig død. Aktuelt er der ikke et standardiseret screeningsprogram for HAVM, hvilket kan bevirke forsinket relevant behandling. Den medicinske behandling er blevet betydeligt bedre over de senere år, dog er den eneste kurative behandling fortsat LTX. Med denne artikel er det håbet, at opmærksomheden på HHT øges, samt at man som kliniker er opmærksom på, at patienter med HHT kan få symptomer på hjertesvigt som led i deres HHT. Ligeledes bør man være opmærksom på patienter med nyopstået hjertesvigt, cirroselignende symptomer eller biliære komplikationer, hyppige næseblødninger og familiærhistorik med næseblødninger og telangiektasier. Disse patienter kan have HHT og bør henvises til relevant udredning.
Korrespondance Pernille Darre Haahr. E-mail: Pernille.Darre.Haahr3@rsyd.dk
Antaget 8. februar 2023
Publiceret på ugeskriftet.dk 27. marts 2023
Interessekonflikter Der er anført potentielle interessekonflikter. Forfatternes ICMJE-formularer er tilgængelige sammen med artiklen på ugeskriftet.dk
Referencer findes i artiklen publiceret på ugeskriftet.dk
Artikelreference Ugeskr Læger 2023;185:V12220752
Pernille Darre Haahr, Jens Kjeldsen, Mikael Kjær Poulsen, Anette Drøhse Kjeldsen & Annette Dam Fialla
Ugeskr Læger 2023;185:V12220752
Hereditary haemorrhagic telangiectasia is a genetic disease, causing abnormal formations of blood vessels in skin, mucus membranes, lungs, liver, and brain. In the liver, the disease results in shunting of blood, bypassing the capillary bed. Recent studies have shown that the prevalence of liver shunts are more frequent than previously suggested. The patients present with symptoms related to high-output cardiac failure causing dyspnoea and oedema. Liver shunts can be shown using CT-scans and ultrasonography. The only curable treatment is a liver transplant; however, it is the last treatment option, as argued in this review.