Statusartikel
Medfødt hørenedsættelse hos børn
Bringes med forældres tilladelse
13. okt. 2025
12 min.
Neonatal hørescreening giver mulighed for identifikation af medfødt høretab.
Høretabets specifikke årsag kan i mange tilfælde påvises ved genetisk udredning.
Kendskab til høretabets årsag har betydning for den relevante prognose og behandling, herunder mulighed for specifik genterapi i fremtiden.
Hørenedsættelse forekommer hos 1-2 pr. 1.000 levendefødte børn [1, 2]. Arvelige årsager til hørenedsættelse udvides hele tiden og udgør > 50% af alle medfødte høretab [3]. Der er derfor tiltagende fokus på, at denne viden kan anvendes til specialiseret behandling og opfølgning af børn med arvelig hørenedsættelse. I Danmark tilbydes alle nyfødte hørescreening, hvilket udføres på landets fødesteder. Ved mistanke om høretab eller ved kendskab til arvelig hørenedsættelse i familien skal barnet henvises til videre udredning. Udredning af spædbørn med høretab er en specialfunktion for de audiologiske afdelinger i Danmark, og den skal være gennemført inden tremånedersalderen, for at behandling af et høretab kan etableres af hensyn til barnets sproglige og kognitive udvikling, der er afhængig af høresansen. Identifikation af en specifik arvelig årsag til hørenedsættelse i forbindelse med udredningen kan danne grundlag for en specialiseret og individuel behandling og opfølgning.
Den neonatale hørescreening blev indført i Danmark i 2005 for at identificere medfødte hørenedsættelser og iværksætte rettidig og korrekt behandling af hørenedsættelsen [4, 5] samt at forebygge udviklingsmæssige forsinkelser relateret til høretabet [6, 7]. Tidligere anvendtes BOEL-testen (»Blikket Orienteret Efter Lyd«) i syv-nimånedersalderen hos sundhedsplejersken som hørescreening, men BOEL-testen er ikke specifik eller sensitiv nok til at identificere hørenedsættelse hos børn. I dag benyttes screeningsmetoderne Transient Evoked Otoacoustic Emissions (TEOAE) eller Distortion Product Otoacoustic Emissions (DPOAE) og Automatic Auditory Brainstem Response (AABR) til identifikation af medfødt høretab.
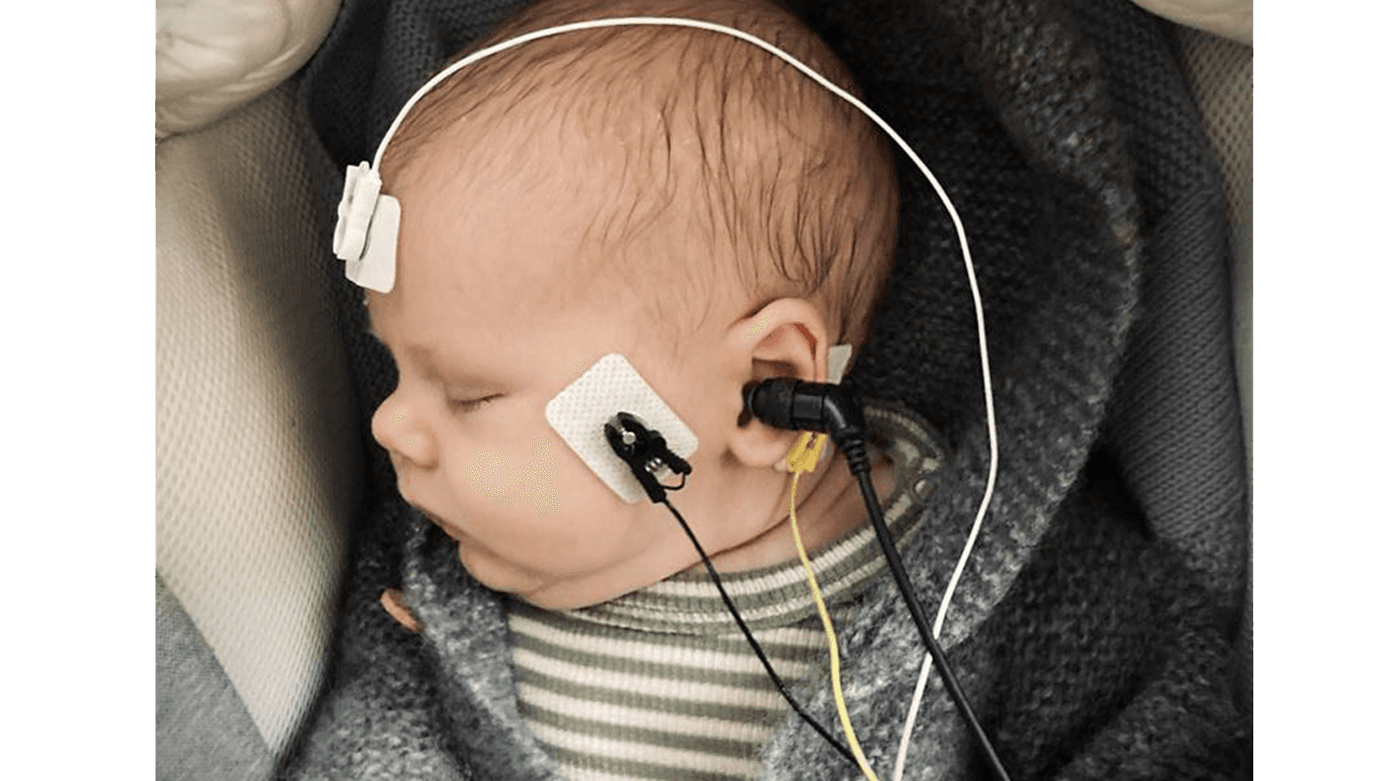
TEOAE er hurtigere, men mindre frekvensspecifik end DPOAE, og testene anvendes til at påvise fungerende ydre hårceller i cochlea, der ved stimulation returnerer en afledt lyd. Da langt de fleste høretab involverer tab af ydre hårceller i cochlea, er testen anvendelig til at påvise moderat til svær hørenedsættelse [8, 9]. Sensitivitet og specificitet er generelt set > 80% ved TEOAE [10], og i et tidligere dansk studie fandt man, at normal hørelse blev klassificeret korrekt i 89% af tilfældene ved brug af TEOAE [11]. Ved AABR sendes kliklyde ind i øregangen, som opfanges i cochlea. Det genererer et fysiologisk respons i form af et aktionspotentiale gennem hørenerven og videre op gennem hjernestammen, hvorfra det neurofysiologiske respons opfanges af elektroder sat på hovedet af barnet (Figur 1). Ved et veldefineret nerverespons efter det givne stimulus ved man, at der er et fungerende høresystem højt op i hjernestammen. AABR identificerer høretab som følge af skade på ydre og indre hårceller samt de sjældne retrokokleære høretab involverende synapsen, hørenerven og hjernestammen. AABR kan påvise normal hørelse korrekt i 96% af tilfældene [11]. AABR benyttes til primær hørescreening alene eller sammen med TEOAE og ved rescreening af børn, der ikke består primær hørescreening, eller børn, som har forøget risiko for at udvikle et høretab (Tabel 1).
Ved fortsat mistanke om hørenedsættelse ved rehørescreeningen fastlægges høretærskler med objektive høreundersøgelser, da det ikke er muligt at fastlægge høretærskler subjektivt hos børn under fem-seksmånedersalderen. Høretærskler fastlægges med Auditory Brainstem Response (ABR) og Automatic Steady State Response (ASSR) [12], mens barnet sover eller er i generel anæstesi.
ABR er registrering af hjernestammesvar efter lydstimulation. Undersøgelsen kan vise, om der kan være tale om et konduktivt høretab, sensorineuralt høretab, blandet høretab eller retrokokleært høretab. Undersøgelsen kan også anvendes til at stille tidlig indikation for cochleaimplantat (CI) til behandling af svær hørenedsættelse hos børn [13] eller til at fastlægge høretærsklen, så høreapparater kan indstilles korrekt. Fastlæggelse af høretærskler i frekvensområdet 500 Hz til 4 kHz kan også ske ved brug af ASSR.
I Europa skyldes omtrent 70% af prælinguale høretab genetiske årsager, og omkring 30% skyldes erhvervede/miljømæssige årsager [14, 15] (Figur 2). Kongenit cytomegalovirus (cCMV)-infektion er den hyppigst diagnosticerede årsag til medfødt ikkearvelig hørenedsættelse. Sygdommen kan være symptomatisk eller asymptomatisk ved fødslen og medføre en markant øget risiko for hastig progression af hørenedsættelsen samt tab af balancefunktion, hvilket bidrager til, at to-tre børn ud af 1.000 levendefødte børn i fireårsalderen har høretab [2, 16].
Der er evidens for effekt af antiviral behandling inden for de første fire leveuger, og derfor er udredning og tidlig diagnosticering ved mistanke om cCMV-infektion afgørende for at minimere hørenedsættelsen [17]. Diagnosen kan stilles ved påvisning af cytomegalovirus (CMV)-DNA i urin, men kan også påvises i spyt, blod og cerebrospinalvæske. Ofte diagnosticeres CMV først meget sent ved påvisning af CMV-infektion efter undersøgelse af blod fra PKU-kort.
Andelen af børn med hørenedsættelse med genetisk baggrund er stigende, i takt med at erhvervede årsager kan behandles og forebygges mere effektivt [18, 19]. Hørenedsættelse med genetisk baggrund kan underinddeles i syndromal og nonsyndromal hørenedsættelse, afhængigt af om andre symptomer eller misdannelser kan påvises. Den nonsyndromale gruppe er størst og udgør ca. 70%. Nonsyndromal eller isoleret hørenedsættelse er med udtalt heterogen genetisk baggrund, hvor der er beskrevet > 150 gener som monogenetisk forårsagende. En opdateret liste kan ses på Hereditary Hearing Loss Homepage [20]. Autosomal recessiv arvegang ses hyppigst (hos omtrent 80%), men autosomal dominant X-bunden arvegang og mere sjældent mitokondriel arvegang ses også. Syndromal hørenedsættelse er beskrevet hos > 400 forskellige syndromer, ligeledes med forskellig arvegang [18, 19]. Syndromale hørenedsættelser håndteres i tværfagligt samarbejde typisk med øjen- og børnelæger.
Ved mistanke om syndromale ensidige og ved alle dobbeltsidige sensorineurale hørenedsættelser med debut før 18-årsalderen tilbydes genetisk udredning. Tidligere undersøgte man på grund af metodemæssige begrænsninger og manglende klinisk konsekvens kun for enkelte og hyppige genetiske årsager til hørenedsættelse. I dag er der adgang til parallel sekventering, dvs. next generation sequencing (NGS), og der er national konsensus om at tilbyde genetisk udredning til patienter med bilateral hørenedsættelse og debut i barnealderen. Typisk undersøges med et genpanel på blodprøve fra patienten, som indebærer samtidig screening af et stort antal relevante gener, omkring 190 gener ved isoleret høretab. En sådan panelundersøgelse kan udføres på basis af flere analysemetoder såsom NGS, exomsekventering eller genomsekventering.
Ved syndromalt høretab vil der også ske årsagsafklaring af de øvrige symptomer hos patienten, som kan være intellektuelt handikap, misdannelser af ydre øre, nyremisdannelser etc. Strategien for udredningen er individuel og kan være panelundersøgelse eller mere udvidet genomanalyse, hvor hele genomet undersøges. Ved helgenomanalyse vil der suppleres med udredning af forældrene, en såkaldt trioanalyse, for at lette fortolkningen af påviste varianter.
Patogene varianter i GJB2 repræsenterer knapt en tredjedel af de genetisk betingede hørenedsættelser hos børn og er derfor den hyppigste form for nonsyndromal hørenedsættelse i Danmark [21] og i resten af verden [22]. GJB2 koder for et transmembranøst gap-junction protein, connexin-26, der sikrer, at kalium recirkuleres fra hårcellerne og tilbage til endolymfen i cochlea, så det endolymfatiske potentiale opretholdes (Figur 3). Kaliumioner er nødvendige for depolarisering af de ydre hårceller, så aktionspotentialet genereres og fører signalet til hjernen. Connexin-26-associeret hørenedsættelse manifesteres oftest som en medfødt svær døvhed, men der fremkommer også flere varianter i GJB2 medførende moderate og progredierende høretab [22], formentlig fordi vi i dag genetisk tester flere med milde til moderate hørenedsættelser. De fleste patienter behandles med CI tidligt, når der er stillet en diagnose med svært høretab, og det gør, at børnene potentielt udvikler sprog på lige fod med normalthørende børn [23].
STRC(-CATSPER2)-varianter diagnosticeres hyppigt som årsag til arveligt betinget høretab, og denne gruppe er den næsthyppigste monogenetiske årsag til nonsyndromal sensorineural hørenedsættelse [24]. STRC koder for proteinet stereocilin, som fungerer som krydsbindinger på toppen af sterocilierne og er vigtig for stabiliteten i de ydre hårceller i cochlea (Figur 3). Fænotypisk ses et ofte symmetrisk, stabilt, mildt til moderat høretab, som typisk kan rehabiliteres med konventionelle høreapparater, og oftest er høretabet ikke så udtalt, at CI kommer på tale. Er der tale om en STRC-deletion, som også involverer CATSPER2 (som er relevant for motilitet af spermatozoer), kan det medføre nedsat fertilitet hos drengebørn [25].
Pendreds syndrom forårsaget af biallelle SLC26A4-mutationer er associeret med forstørret vestibulær akvædukt og endolymfatisk sækanomali samt en mindre misdannelse i cochlea (incomplete partition type 2/IP-2), som skyldes manglen på proteinet pendrin under dannelsen af indre øre i forstertilværelsen (Figur 2). Derudover ses typisk eutyroid struma, der oftest udvikles før og i teenagealderen. Langt de fleste børn med biallel SLC26A4-mutationer vil progrediere til svær hørenedsættelse inden for 3.-6. leveår [26]. Genetisk udredning er således nødvendigt prognostisk, eftersom børnene kan have et mildt eller moderat høretab ved fødslen og endog kan bestå den neonatale screening på ét øre. Børnene kan opleve fluktuationer i hørelsen, som påvirker børnenes sproglige udvikling og gør høretest vanskelige. Genetisk udredning er derfor afgørende i forhold til stillingtagen til tidlig CI (som minimum ensidigt), der er væsentligt ved fluktuerende hørenedsættelse for netop at sikre børnenes hørelse igennem sprogudviklingen.
Otoferlin (OTOF)-varianter er associeret med hørenedsættelse og auditiv neuropati, der diagnosticeres ved at påvise DPOAE eller TEOAE og fravær af et ABR-signal. Således vil hørescreening uden brug af AABR risikere ikke at identificere høretab forårsaget af defekt OTOF. OTOF-mutationer er sjældne, men banebrydende for anvendelse af genterapi til monogenetisk betinget hørenedsættelse. OTOF koder for proteinet otoferlin, der faciliterer præsynaptisk signaltransmission fra indre hårceller til synapsen mod hørenerven (Figur 3), hvilket er årsagen til, at otoferlin er en oplagt kandidat for behandling med genterapi. Simultane kliniske studier i USA, Taiwan og Storbritannien viser foreløbige lovende resultater [27], og to publicerede studier fra Kina rapporterer om genskabelse af hørelse efter en intrakokleært administreret modificeret adenoassocieret viral vektor via det runde vindues niche [28, 29].
Effektiv hørescreening med brug af AABR og DPOAE/TEOAE vil identificere de fleste høretab således, at karakteristik af den genetiske årsag til høretabet efterfølgende kan udføres for at muliggøre specifik behandling af børn med medfødte hørenedsættelser. Hvor vi i dag anvender høreapparater til de moderate hørenedsættelser og CI til de svære hørenedsættelser, vil vi formentlig se stadigt flere genterapeutiske muligheder for at behandle specifikke monogenetiske årsager til medfødt hørenedsættelse. Det bliver derfor altafgørende, at årsagen til de genetiske høretab udredes, når der potentielt er mulighed for, at hørenedsættelsen kan mindskes eller helt elimineres ved genterapi i fremtiden. Fund af genetiske varianter, som man endnu ikke kender betydningen af, rummer dog en udfordring i forhold til at forklare forældre til børn med høretab, hvad de kan forvente af fremtiden.
Korrespondance Jesper Hvass Schmidt. E-mail: jesper.schmidt@rsyd.dk
Antaget 24. juli 2025
Publiceret på ugeskriftet.dk 13. oktober 2025
Interessekonflikter KM oplyser økonomisk støtte fra eller interesse i Advanced Bionics og Cochlear. BE oplyser økonomisk støtte fra eller interesse i Cochlear. JHS oplyser økonomisk støtte fra eller interesse i Cochlear, beta.health og Genmab A/S. Alle forfattere har indsendt ICMJE Form for Disclosure of Potential Conflicts of Interest. Disse er tilgængelige sammen med artiklen på ugeskriftet.dk
Referencer findes i artiklen publiceret på ugeskriftet.dk
Artikelreference Ugeskr Læger 2025;187:V03250237
doi 10.61409/V03250237
Open Access under Creative Commons License CC BY-NC-ND 4.0
The identification of congenital hearing loss using Transient Evoked Otoacoustic Emissions and Automatic Auditory Brainstem Response in a newborn hearing screening program is crucial for initiating early rehabilitation with hearing aids or cochlear implants. Specific genetic causes, such as Pendred syndrome, connexin-26, stereocilin, and otoferlin-associated deafness, can be identified today using gene panels. Specifically, for otoferlin-associated deafness, it may be possible to offer gene therapy as a novel treatment for this specific genetic type of hearing loss.